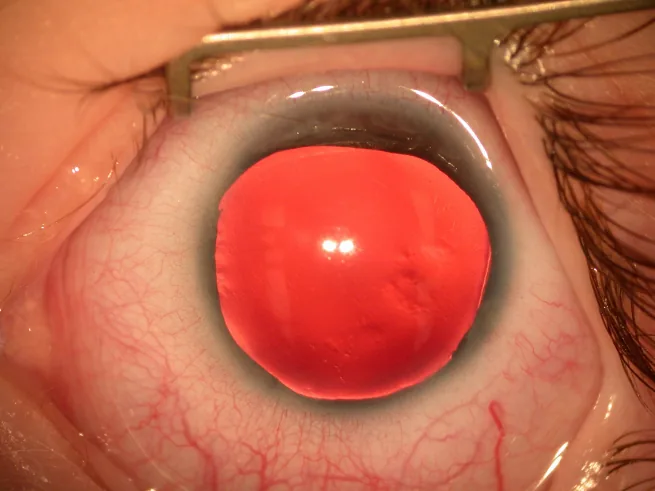
L’aniridia è una condizione in cui l’iride è completamente o parzialmente assente a causa di una predisposizione congenita. Sebbene sia chiamata “aniridia”, spesso rimane un residuo della radice dell’iride nella parte più periferica dell’angolo camerulare.
Nel 2017 è stata riconosciuta come malattia rara designata ai sensi della legge sulle malattie rare del Ministero della Salute, del Lavoro e del Welfare 1). I pazienti diagnosticati con questa malattia rara designata e classificati con un grado di gravità pari o superiore a III hanno diritto a un contributo per le spese mediche, con un limite massimo di auto-copertura basato sul reddito 2).
Circa 2/3 dei casi (ereditarietà autosomica dominante)
Sporadico
Circa 1/3 dei casi
Associazione con tumore di Wilms (casi sporadici)
Circa 30% (sindrome WAGR)3)
Studi epidemiologici in Svezia e Norvegia riportano una prevalenza di circa 1 su 90.000 persone3). Una valutazione oftalmologica dettagliata di 43 casi con mutazioni del gene PAX6 ha mostrato che il grado di anomalia dell’iride varia a seconda del tipo di mutazione3).
QL'aniridia è ereditaria?
A
Circa 2/3 dei casi sono ereditari con trasmissione autosomica dominante, con una probabilità del 50% di trasmettere la malattia da un genitore affetto al figlio. Il restante 1/3 è sporadico, senza storia familiare. Nei casi sporadici, esiste il rischio di sindrome WAGR, che include il tumore di Wilms (tumore renale), pertanto si raccomanda il test genetico per i geni PAX6 e WT1.
Law SK, et al. Asymmetric phenotype of Axenfeld-Rieger anomaly and aniridia associated with a novel PITX2 mutation. Mol Vis. 2011. Figure 2. PMCID: PMC3102021. License: CC BY.
Fotografia con lampada a fessura del segmento anteriore: l’iride è quasi completamente assente, con solo un sottile residuo di iride visibile alla periferia. Mostra direttamente i segni clinici tipici dell’aniridia, adatta per descrivere i principali sintomi e segni clinici.
A causa dell’assenza o dell’incompletezza dell’iride, la pupilla non funziona e non può regolare la quantità di luce che entra nell’occhio. Ciò provoca una forte fotofobia. Inoltre, la scarsa fissazione dovuta all’ipoplasia maculare è spesso la causa principale del nistagmo orizzontale che si manifesta precocemente dopo la nascita.
Fotofobia: incapacità di regolare la luce a causa dell’iride → forte abbagliamento
Nistagmo (nistagmo orizzontale): scarsa fissazione dovuta a ipoplasia maculare. Compare precocemente dopo la nascita
Riduzione dell’acuità visiva: fattori combinati di ipoplasia maculare, cataratta, glaucoma e deficit limbico corneale (LSCD)
Anomalia dell’iride: vari gradi, da atrofia parziale a completa assenza
Ipoplasia maculare: presente in quasi tutti i casi. Scomparsa della fovea e pigmento maculare poco definito. Principale fattore limitante dell’acuità visiva
Nistagmo: prevalentemente orizzontale. Causato da ipoplasia maculare
Strabismo: compare a causa della scarsa acuità visiva
Complicanze acquisite (insorgono con la crescita)
Cataratta: presente in circa l’80% dei casi. Insorge nel 50-85% entro i 20 anni
Glaucoma: presente nel 50-75% dei casi. Raro nell’infanzia, progressivo in età giovanile
Deficienza limbica delle cellule staminali (LSCD): spesso normale in età infantile, ma con la crescita progrediscono opacità stromale corneale e panno vascolare
Riepilogo della frequenza delle complicanze oculari
Il gene PAX6 è espresso non solo nei tessuti oculari, ma anche nel sistema nervoso centrale, nelle isole di Langerhans del pancreas e nell’epitelio olfattivo. L’ipoplasia di questi tessuti può causare diverse complicanze extraoculari1).
Agenesia del corpo calloso, epilessia, disturbi delle funzioni cerebrali superiori
Anosmia
Intolleranza al glucosio
Sindrome WAGR (circa il 30% dei casi sporadici): tumore di Wilms, aniridia, anomalie genitourinarie, ritardo mentale3)
QQuanto si vede con l'aniridia?
A
La prognosi visiva è generalmente sfavorevole, spesso intorno a 0,1. Tuttavia, varia da 0,1 a 0,7 a seconda del grado di ipoplasia maculare e della presenza di complicanze. L’ipoplasia maculare, per la quale attualmente non esiste una terapia efficace, è il principale fattore limitante dell’acuità visiva. Una corretta correzione refrattiva e la riabilitazione per ipovisione possono migliorare la qualità della vita quotidiana.
L’aniridia è causata dalla perdita di funzione di un allele (aploinsufficienza) del gene PAX6, situato sul braccio corto del cromosoma 11 (11p13). Ciò è dovuto alla riduzione della metà del dosaggio genico funzionale. Si ritiene che l’anomalia di entrambi gli alleli sia letale in fase embrionale1).
PAX6 è un gene master control che codifica per un fattore di trascrizione responsabile della differenziazione degli organi in fase embrionale, regolando vari altri fattori di trascrizione. Le anomalie di PAX6 causano diverse malformazioni congenite dell’intero bulbo oculare (aniridia, anomalia di Peters, ipoplasia maculare, ecc.).
I tipi di mutazione genica sono spesso mutazioni di tipo PTC (codone di terminazione prematuro) come nonsenso e frameshift, ma sono state riportate anche mutazioni missenso1). Nell’aniridia isolata, l’analisi di sequenziamento rileva mutazioni di PAX6 in circa l’85% dei casi2).
Il gene PAX6 è adiacente al gene oncosoppressore WT1 sul cromosoma 11p13. Nei casi sporadici, una delezione genica contigua può causare la sindrome WAGR, caratterizzata da tumore di Wilms, aniridia, anomalie genitourinarie e ritardo mentale3). In circa il 30% dei casi sporadici, il tumore di Wilms si sviluppa precocemente e bilateralmente entro i 5 anni di età.
Mutazione PAX6 positiva senza delezione di WT1 → si può presumere l’assenza di sindrome WAGR2)
Il test genetico combina il sequenziamento del DNA con la rilevazione di anomalie strutturali genomiche tramite MLPA/CMA2)
Nei casi sporadici sospetti per sindrome WAGR, è raccomandato il test genetico 2)
QDovrei sottopormi al test genetico per l'aniridia?
A
Il test genetico per PAX6 è necessario per confermare la diagnosi definitiva, e nei casi sporadici è raccomandato il test genetico per PAX6 e WT1 per valutare il rischio di tumore di Wilms. È importante eseguire il test combinando sequenziamento del DNA e MLPA/CMA, sotto appropriata consulenza genetica.
Fotofobia (in base al grado di difetto dell’iride)
B. Reperti diagnostici
All’esame con lampada a fessura, anomalie della formazione dell’iride che vanno dall’atrofia parziale all’assenza completa dell’iride (bilaterale nel 60-90% dei casi)
All’esame del fondo oculare e OCT, ipoplasia maculare (fovea poco definita, pigmento maculare e area avascolare foveale indistinti)
Screening per glaucoma. Eseguire regolarmente dall’adolescenza
Ecografia addominale
Screening per tumore di Wilms (casi sporadici, ogni pochi mesi, specialmente fino a 5 anni)
Test genetico
Identificazione di mutazioni del gene PAX6 o delezione della regione 11p13 (necessaria per diagnosi definitiva)
Nei bambini può essere necessario un esame in anestesia generale.
QCome viene diagnosticata l'aniridia?
A
L’esame di base prevede la conferma della malformazione dell’iride con lampada a fessura e la valutazione dell’ipoplasia maculare con OCT. La diagnosi definitiva è possibile con il test genetico per PAX6; nei casi sporadici si ricerca anche il gene WT1. È importante la diagnosi differenziale con atrofia dell’iride erpetica, difetto irideo post-traumatico, coloboma dell’iride, anomalia di Rieger e sindrome ICE.
Attualmente non è possibile intervenire sulla malformazione dell’iride, l’ipoplasia maculare, la microftalmia e il nistagmo; pertanto la gestione si basa sull’osservazione. Il trattamento è mirato a cheratopatia, cataratta, glaucoma, fotofobia e ipovisione2).
Opacità dello stroma corneale: Il miglioramento della funzione visiva ottenuto con il trapianto di cornea è limitato dalle complicanze dell’aniridia2). A lungo termine, la prognosi visiva è spesso sfavorevole a causa del peggioramento del glaucoma e del fallimento del trapianto nel tempo. Il trapianto di cornea a tutto spessore per l’opacità corneale spesso non porta a un miglioramento della vista e si deve prestare attenzione all’alto tasso di rigetto. Nei casi gravi, la decisione di procedere deve essere presa dopo aver valutato attentamente il rapporto tra benefici e rischi.
Deficienza di cellule staminali limbari (LSCD): Si deve considerare il trattamento chirurgico2). Nello specifico, il trapianto di limbo allogenico (KLAL) o il trapianto di epitelio orale coltivato (COMET) possono offrire una certa ricostruzione della superficie oculare3). In caso di concomitante opacità dello stroma corneale, la combinazione con il trapianto di cornea è spesso utile per migliorare la vista2).
Entro i 20 anni, il 50-85% dei pazienti sviluppa la cataratta e si pianifica l’intervento di cataratta in base all’intensità dell’opacità e della fotofobia2).
Difficoltà chirurgica elevata a causa della fragilità del sacco capsulare e della zonula di Zinn
Prestare attenzione al rischio di peggioramento del glaucoma post-operatorio, sindrome da fibrosi anteriore e cheratopatia bollosa2)
Poiché il glaucoma influisce direttamente sulla prognosi visiva, deve essere trattato attivamente2). Si adotta il seguente approccio graduale.
Terapia farmacologica: Riduzione della pressione intraoculare tramite colliri e farmaci orali, prestando attenzione agli effetti collaterali e considerando l’impatto sistemico nei bambini
Chirurgia di ricostruzione del deflusso: Goniotomia e trabeculotomia (da considerare se la terapia farmacologica è inefficace)
Chirurgia dell’impianto per glaucoma: intervento con tubo lungo (richiede certificazione della struttura)
Ciclocoagulazione: ultima risorsa quando altri trattamenti falliscono
La resistenza alla terapia farmacologica è frequente e la chirurgia con shunt tubulare può essere una buona opzione4). Poiché il danno del campo visivo nel glaucoma è irreversibile, il controllo precoce della pressione intraoculare è fondamentale per preservare la funzione visiva.
QCome si tratta il glaucoma nell'aniridia?
A
Inizialmente si ricorre alla terapia farmacologica con colliri e farmaci orali, ma spesso il glaucoma è resistente ai farmaci. Se inefficace, si considera la chirurgia ricostruttiva del deflusso (goniotomia/trabeculotomia), per poi procedere con trabeculectomia o chirurgia con tubo lungo (impianto per glaucoma). L’intervento con tubo lungo richiede la certificazione della struttura. La ciclocoagulazione è l’ultima risorsa quando altri trattamenti falliscono. È indispensabile un monitoraggio regolare della pressione intraoculare.
Le cure per l’ipovisione e la gestione della fotofobia dovrebbero essere introdotte precocemente per preservare la funzione visiva e la qualità della vita2).
Correzione refrattiva: correggere i vizi di rifrazione con occhiali per favorire lo sviluppo visivo il più possibile (di base)
Occhiali schermanti: efficaci per ridurre la fotofobia. Prescritti in caso di fotofobia intensa
Lenti a contatto con iride artificiale: utili sia per migliorare la fotofobia che l’aspetto estetico
Utilizzare ausili visivi come lenti d’ingrandimento, occhiali per ipovedenti e ingranditori per lettura
Il gene PAX6 è un gene di controllo master che codifica per un fattore di trascrizione che regola la differenziazione degli organi durante il periodo embrionale. Si esprime nell’occhio primordiale e coordina vari fattori di trascrizione. La perdita di funzione di un allele di PAX6 (aploinsufficienza) causa anomalie congenite in tutto l’occhio (aniridia, anomalia di Peters, ipoplasia maculare, ecc.).
Le mutazioni di PAX6 sono spesso di tipo PTC (nonsenso, frameshift), ma sono state riportate anche mutazioni missenso1). Gli studi sulla correlazione genotipo-fenotipo mostrano che la gravità dei reperti oftalmici varia a seconda del tipo di mutazione3).
PAX6 si esprime anche al di fuori dell’occhio, nel sistema nervoso centrale, nelle isole di Langerhans del pancreas e nell’epitelio olfattivo; l’ipoplasia di questi tessuti può causare complicanze extraoculari (agenesia del corpo calloso, epilessia, anosmia, intolleranza al glucosio)1).
Patologia ad angolo aperto: aumento della resistenza al deflusso dell’umore acqueo nel trabecolato
Patologia ad angolo chiuso: la radice dell’iride residua nella parte più periferica aderisce al trabecolato, causando una forma di glaucoma ad angolo chiuso
Il glaucoma è raro nell’infanzia e si sviluppa progressivamente in età giovanile con la crescita. Può verificarsi in caso di angolo aperto a causa di una malformazione dell’angolo o in caso di angolo chiuso.
Patologicamente, si osserva una disfunzione delle cellule staminali epiteliali corneali, con anomalie dell’epitelio e della membrana di Bowman, e formazione di panno vascolarizzato. L’ipoplasia delle palisades di Vogt progredisce verso l’invasione del tessuto congiuntivale e la cheratinizzazione1).
La cornea nell’aniridia è più spessa rispetto ai soggetti sani. Spesso la cornea è normale durante l’infanzia, ma con la crescita si sviluppano opacità stromali corneali e LSCD, causando una riduzione dell’acuità visiva. In uno studio monocentrico di 14 anni (738 occhi), l’aniridia era la causa più frequente di LSCD, rappresentando il 30,9% dei casi6).
La prognosi visiva è generalmente sfavorevole, spesso intorno a 0,1
L’ipoplasia maculare non ha un trattamento efficace ed è il principale fattore limitante l’acuità visiva
Il danno del campo visivo da glaucoma è irreversibile; è importante un controllo precoce della pressione intraoculare
Nei casi sporadici, prestare attenzione all’insorgenza precoce del tumore di Wilms entro i 5 anni e continuare regolari ecografie addominali.
Gli studi sulla prognosi a lungo termine riportano che la prognosi visiva è generalmente sfavorevole, ma varia individualmente in base al tipo e alla gravità delle complicanze5).
Con la diffusione del sequenziamento di nuova generazione (NGS), il tasso di rilevamento delle mutazioni PAX6 nell’aniridia isolata è di circa l’85%2). Il microarray cromosomico (CMA) è più sensibile dei test cromosomici tradizionali nel rilevare microdelezioni in 11p13, contribuendo a migliorare l’accuratezza diagnostica della sindrome WAGR2).
Si stanno accumulando dati a lungo termine sul trapianto di mucosa orale coltivata (COMET)2). Per la cheratoprotesi Boston tipo I, a breve termine (17-28,7 mesi) si ottiene un miglioramento visivo nel 65-93% dei casi, ma a 4,5 anni la percentuale scende al 43,5%2).
Prospettive per i dispositivi a iride artificiale e la terapia genica
L’HumanOptics CustomFlex ArtificialIris è un dispositivo a iride artificiale in silicone su misura, utile per ridurre la fotofobia e migliorare l’aspetto, ma non è ancora approvato in Giappone al 2024. La terapia mirata per l’aploinsufficienza di PAX6 è attualmente in fase di ricerca e non ha ancora raggiunto l’applicazione clinica3).
Hingorani M, Hanson I, van Heyningen V. Aniridia. Eur J Hum Genet. 2012 Oct;20(10):1011-1017. doi:10.1038/ejhg.2012.100. PMID:22692063; PMCID:PMC3449076.