Le anomalie dello sviluppo del segmento anteriore (Anterior Segment Developmental Anomalies; ASDA) sono un termine collettivo per i disturbi dello sviluppo che coinvolgono il segmento anteriore dell’occhio — cornea, iride, cristallino e camera anteriore. Sono anche chiamate “disgenesia del segmento anteriore” (Anterior Segment Dysgenesis; ASD).
L’ASDA include le seguenti unità patologiche rappresentative.
Queste malattie sono eterogenee sia dal punto di vista fenotipico che genetico, e si sa che oltre 50 geni sono coinvolti. Le conoscenze genetiche continuano ad espandersi grazie all’analisi dell’esoma e del genoma intero, ma nel 40-75% dei casi il gene causale non è ancora stato identificato. I casi che non possono essere classificati in un fenotipo specifico vengono descritti come “ASD non classificabile (unclassified ASD)”. 1)
L’umore acqueo prodotto dal corpo ciliare dell’iride viene drenato attraverso il trabecolato (trabecular meshwork) nel canale di Schlemm e anche attraverso la via uveosclerale. Nell’ASDA questo processo è spesso compromesso, e il glaucoma secondario è una complicanza comune e importante.
Se è presente solo l’anello embrionale posteriore senza sintomi sistemici, in base al 9° rapporto di consenso della World Glaucoma Association, viene trattato separatamente dall’ARS. 1)
QA che età viene diagnosticata l'anomalia dello sviluppo del segmento anteriore (ASDA)?
A
Varia a seconda della malattia. Il glaucoma congenito primario si manifesta spesso entro il primo anno di vita. La sindrome di Axenfeld-Rieger e l’anomalia di Peters vengono spesso diagnosticate alla nascita. Il glaucoma evolutivo tardivo può manifestarsi fino all’età di 10-20 anni. In tutti i casi, la diagnosi precoce e il trattamento tempestivo sono importanti.
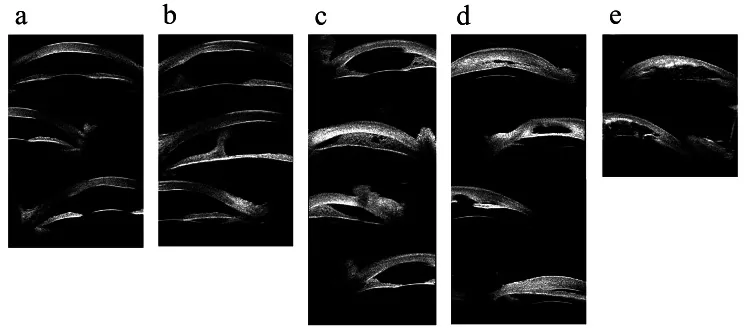
Hong J, et al. Classifications of anterior segment structure of congenital corneal opacity in infants and toddlers by ultrasound biomicroscopy and slit-lamp microscopic photographs: an observational study. BMC Ophthalmol. 2024. Figure 1. PMCID: PMC10804776. License: CC BY.
Immagini di biomicroscopia ultrasonica di (a) opacità corneale, (b) opacità corneale e sinechia anteriore centrale, (c) sinechia iridocorneale periferica inferiore a 180 gradi, (d) sinechia iridocorneale periferica superiore a 180 gradi, (e) opacità corneale con anomalie dell’iride e del cristallino. Corrisponde alle sinechie del segmento anteriore e alle opacità corneali trattate nella sezione “2. Sintomi principali e reperti clinici”.
Nell’infanzia, i seguenti sintomi associati all’aumento della pressione intraoculare sono spesso osservati come sintomi iniziali.
Lacrimazione (eccessiva lacrimazione): si verifica come risultato dell’irritazione associata all’edema epiteliale corneale causato dall’aumento della pressione intraoculare.
Fotofobia (ipersensibilità alla luce): sintomo che riflette l’irritazione corneale.
Blefarospasmo: compare con lo stesso meccanismo della lacrimazione e della fotofobia.
Nei bambini più grandi e negli adulti, nella forma tardiva si possono avvertire offuscamento della vista e diminuzione dell’acuità visiva già in età relativamente giovane. Se la pressione intraoculare è molto elevata, possono manifestarsi sintomi come affaticamento visivo e mal di testa. Nell’aniridia, i pazienti possono lamentare fotofobia (cecità diurna).
L’ingrandimento del bulbo oculare (aumento del diametro corneale) e l’opacità corneale sono spesso notati dai genitori e portano alla visita medica.
L’ASDA presenta reperti caratteristici per ciascuna malattia. Di seguito sono riportati i principali reperti delle unità patologiche rappresentative.
Anello di Schwalbe prominente e strie di Axenfeld
Anello di Schwalbe posteriore (PE): linea di Schwalbe anteriormente spostata e ispessita. Alla lampada a fessura si osserva come una linea grigio-biancastra concentrica all’interno del limbo corneale.
Anomalia di Axenfeld: presenza di aderenze a corda del tessuto irideo periferico all’anello di Schwalbe posteriore.
Anomalia di Rieger: oltre a quanto sopra, presenta deviazione pupillare, ectropion uveale e pseudopolicoria dovuti a ipoplasia dello stroma irideo. Ereditarietà autosomica dominante. Nel 50-60% dei casi si associa a glaucoma.
Anomalia di Peters
Opacità corneale centrale: reperto essenziale per la diagnosi. Riflette la perdita dell’endotelio corneale, della membrana di Descemet e dello stroma corneale.
Tipo 1: solo difetto della superficie posteriore della cornea e opacità corneale.
Tipo 2: con sinechie dell’iride.
Tipo 3: associato a spostamento anteriore del cristallino e cataratta. Circa l’80% dei casi è bilaterale. Glaucoma presente nel 50-70% dei casi.
Aniridia
Ipoplasia dell’iride: il difetto riguarda principalmente la porzione posteriore dell’iride. Può essere associata a ipoplasia maculare, ipoplasia del nervo ottico e glaucoma.
Cheratopatia associata ad aniridia (AAK) : l’incidenza riportata varia dal 20% a oltre l’80%. Si tratta di un’opacità corneale progressiva dovuta a insufficienza delle cellule staminali del limbo (LSCD), che progredisce per tutta la vita. 2)
Sindrome WAGR: si verifica quando il gene PAX6 e il gene WT1 adiacente sono mutati. Include tumore di Wilms, aniridia, anomalie genitourinarie e ritardo dello sviluppo psicomotorio. 3)
Tipo di anomalia corneale
Megacornea: diametro corneale ≥13 mm (neonati ≥12 mm). Di solito, pressione intraoculare e densità delle cellule endoteliali sono normali. Prevalentemente eredità recessiva legata all’X.
Scleralizzazione corneale: tessuto sclerale opaco invade la cornea periferica. Il confine tra sclera e cornea è indistinto, con invasione vascolare.
CHED: edema corneale bilaterale simmetrico che compare dalla nascita fino a 1-2 anni di età. Non associato a aumento della pressione intraoculare. Ereditarietà autosomica recessiva.
Di seguito sono riportati i reperti che si aggiungono in caso di glaucoma secondario complicato.
Aumento della pressione intraoculare: può presentarsi con ipertensione oculare (circa 30-50 mmHg).
Aumento del diametro corneale (buftalmo) : dovuto alla distensione del rivestimento oculare. Se supera i 12,0 mm subito dopo la nascita, sospettare un glaucoma congenito.
Linee di Haab: opacità lineari permanenti che rimangono nel sito di rottura della membrana di Descemet.
Aumento dell’escavazione della papilla ottica: nei neonati e nei bambini piccoli, un rapporto C/D superiore a 0,3 fa sospettare un glaucoma. Anche una differenza tra i due occhi superiore a 0,2 è un segno sospetto.
QNella sindrome di Axenfeld-Rieger, in quale percentuale si sviluppa il glaucoma?
A
Si stima che il glaucoma si sviluppi nel 50-60% dei casi (in alcuni rapporti fino al 50-75%), con un’alta frequenza. 3) La trasmissione è autosomica dominante. I casi con sintomi sistemici (anomalie dentali, anomalie facciali, anomalie ipofisarie, ecc.) sono chiamati sindrome di Rieger. Si raccomanda lo screening del glaucoma nei familiari.
La causa principale dell’ASDA è un’anomalia genetica, con diversi geni e modalità di trasmissione coinvolti a seconda della malattia. Di seguito sono elencati i geni responsabili delle principali malattie.
Inoltre, sono stati riportati casi di glaucoma congenito associato ad anomalie genetiche come PAX6, PITX2 e FOXC1. La correlazione tra genotipo e fenotipo è varia, e anche all’interno di una stessa famiglia con la stessa anomalia genetica, il fenotipo può essere diverso.
La maggior parte dei casi di glaucoma congenito ad esordio precoce (glaucoma congenito primario) sono sporadici, ma circa il 10% segue un modello di ereditarietà autosomica recessiva. Esiste anche una teoria che suggerisca un’ereditarietà multifattoriale.
Le cellule della cresta neurale (neural crest cells) svolgono un ruolo centrale nella formazione del segmento anteriore dell’occhio. Le cellule trabecolari derivano dalla cresta neurale, mentre il tessuto connettivo paracanalicolare deriva dalle cellule endoteliali vascolari. Il punto di contatto tra questi tessuti di diversa origine presenta la massima resistenza al deflusso dell’umore acqueo. L’ARS, l’anomalia di Peters e la sindrome da ectropion uveale congenito sono tutte considerate anomalie congenite causate da un’alterata migrazione delle cellule della cresta neurale.
Uno studio su larga scala in Corea ha mostrato che un aumento dell’esposizione materna al PM2.5 (particolato fine) nei tre mesi precedenti il concepimento e durante il primo e secondo trimestre di gravidanza è associato a un rischio maggiore di ASDA nei figli.
La diagnosi di ASDA è principalmente clinica. Nei bambini di età inferiore o uguale a 5 anni, gli esami spesso richiedono l’esecuzione in anestesia generale o sotto sedazione.
Esame con lampada a fessura: valuta il grado e la sede dell’opacità corneale, la presenza della linea di Haab, la profondità della camera anteriore, le anomalie dell’iride (anello di Schwalbe posteriore, adesione dell’iride alla linea di Schwalbe) e le anomalie del cristallino. Verifica la presenza di anello di Schwalbe posteriore, anomalie dell’iride (ARS) e cataratta (anomalia di Peters).
Misurazione del diametro corneale: misurare il diametro orizzontale e verticale con un calibro. Il range normale nei neonati è 9,5-10,5 mm. Se supera i 12,0 mm subito dopo la nascita, sospettare un glaucoma congenito.
Esame dell’angolo: si utilizza una lampada a fessura portatile e un gonioscopio diretto come la lente di Koeppe. Si valutano l’inserzione alta dell’iride, l’adesione dell’iride alla linea di Schwalbe (reperto ARS) e l’aumento della larghezza del trabecolato.
Esame del fondo oculare: osservazione dell’escavazione della papilla ottica. Nei neonati e nei bambini piccoli, un rapporto C/D ≥ 0,3 fa sospettare un glaucoma. La riduzione dell’escavazione papillare dopo la diminuzione della pressione intraoculare è un segno di buon controllo pressorio.
Microscopio ecografico biomicroscopico (UBM): utile nei casi in cui l’opacità corneale rende difficile la visualizzazione dell’angolo. Aiuta anche a valutare il grado di anomalia dell’angolo e a stimare la prognosi della chirurgia di ricostruzione del deflusso.
OCT del segmento anteriore (AS-OCT): come esame complementare, consente una valutazione non invasiva della struttura dell’angolo e della cornea, ma non sostituisce la gonioscopia nella diagnosi. 3)
Esame del campo visivo: essenziale per la diagnosi di neuropatia ottica glaucomatosa pediatrica. Nei bambini di età inferiore a 5 anni è difficile anche per un esaminatore esperto; l’esame del campo visivo dinamico è più facile da eseguire.
Secondo la quarta edizione delle Linee guida per la diagnosi e il trattamento del glaucoma della Società giapponese del glaucoma, il glaucoma pediatrico viene diagnosticato quando sono soddisfatti due o più dei seguenti criteri.
Pressione intraoculare > 21 mmHg
Progressione dell’aumento del rapporto C/D, asimmetria del rapporto C/D ≥ 0.2, assottigliamento del bordo neuroretinico
Reperti corneali: linee di Haab, o nei neonati diametro corneale ≥11 mm, nei bambini <1 anno ≥12 mm, a tutte le età ≥13 mm
Progressione della miopia dovuta a un allungamento dell’asse oculare che supera lo sviluppo normale
Difetto del campo visivo riproducibile coerente con la neuropatia ottica glaucomatosa
Trauma da parto con forcipe: opacità lineare unilaterale, a decorso verticale o obliquo.
Malattie metaboliche come la mucopolisaccaridosi congenita e la cistinuria: la valutazione dei sintomi sistemici è importante per la diagnosi differenziale.
I gruppi di malattie incluse nell’ASD (anomalia di Axenfeld-Rieger, anomalia di Peters, aniridia, distrofia polimorfa posteriore, microftalmia, microcornea, ecc.) devono essere considerati reciprocamente nella diagnosi differenziale. 3)
QL'anello embrionale posteriore (posterior embryotoxon) da solo può causare glaucoma?
A
Un anello embriotossico posteriore isolato (senza sintomi sistemici) si distingue dalla SRA, ma è anche uno dei segni associati alla SRA. L’anello embriotossico posteriore può essere osservato anche in occhi sani e di per sé non indica necessariamente un rischio di glaucoma. Tuttavia, se associato ad altre malattie come la sindrome di Alagille, è necessario monitorare la pressione intraoculare.
La terapia farmacologica è un trattamento ausiliario volto ad abbassare la pressione intraoculare a breve termine prima dell’intervento chirurgico e a controllarla dopo l’operazione. La scelta dei farmaci è sostanzialmente la stessa del glaucoma ad angolo aperto dell’adulto. Tuttavia, i beta-bloccanti richiedono cautela in caso di asma bronchiale e bradicardia, e nei neonati sono stati riportati casi di apnea. È possibile anche la somministrazione orale di acetazolamide (5-10 mg/kg ogni 6-8 ore).
Il glaucoma congenito ad esordio precoce richiede fondamentalmente un trattamento chirurgico. La terapia farmacologica ha un ruolo di supporto.
Goniotomia: è indicata come primo intervento nei casi con lieve opacità corneale. Presenta il vantaggio di non traumatizzare la congiuntiva. Utilizzando una lente di Barkan o di Swan-Jacob, si raschia la superficie del trabecolato corneosclerale con un coltello per goniotomia.
Trabeculotomia: può essere eseguita indipendentemente dalla presenza di opacità corneale. Viene eseguita anche come intervento aggiuntivo quando la goniotomia non è sufficientemente efficace.
Trabeculectomia e chirurgia con tubo di drenaggio: opzioni in caso di inefficacia della chirurgia dell’angolo. Nell’ARS, si sceglie la chirurgia dell’angolo se l’angolo è aperto e la copertura del trabecolato da sinechie anteriori periferiche non è estesa, ma il tasso di successo è inferiore rispetto al PCG. Nei casi in cui la chirurgia dell’angolo è inefficace, la trabeculectomia o la chirurgia con tubo di drenaggio con placca possono essere la prima scelta. 4)
Nell’anomalia di Peters, il trattamento segue quello del glaucoma congenito primario (PCG), ma solo circa un terzo degli occhi operati raggiunge una buona pressione intraoculare postoperatoria, e molti casi hanno una prognosi sfavorevole. A causa delle anomalie corneali associate, spesso è difficile ottenere una visione utile. 4)
Anomalia di Peters: nei casi lievi, l’opacità corneale spesso diminuisce gradualmente. Se la pressione intraoculare è normale, si osserva spesso un certo miglioramento e, poiché il decorso dopo il trapianto di cornea è sfavorevole, il trapianto di cornea nell’infanzia di solito non viene eseguito. Molti casi sono resistenti al trattamento farmacologico del glaucoma e, anche dopo un intervento di ricostruzione della via di deflusso, sono difficili da controllare e hanno una prognosi sfavorevole.
CHED: In caso di insufficienza endoteliale corneale, il trapianto di cornea (incluso il trapianto endoteliale) può essere indicato.
Cornea sclerale: può essere associata ad altre sindromi ASD; nei casi gravi può essere indicato il trapianto di cornea.
Fotofobia e affaticamento visivo: considerare l’uso di occhiali schermanti e la prescrizione di lenti a contatto cosmetiche.
Cheretopatia associata ad aniridia (AAK): per l’opacità corneale progressiva, si considera il trapianto di cellule staminali del limbus corneale.
Screening per tumore di Wilms: in caso di aniridia sporadica, si raccomanda di indirizzare il paziente a un pediatra e di effettuare lo screening fino all’età di 6 anni. 4)
Anche se la pressione intraoculare diminuisce, spesso è necessario il trattamento dell’ambliopia. Poiché l’aneisometropia, l’astigmatismo irregolare, l’opacità corneale e le strie di Haab possono causare ambliopia, è necessario continuare a monitorare l’acuità visiva e la refrazione parallelamente alla misurazione della pressione intraoculare. La progressione della miopia e l’allungamento della lunghezza assiale suggeriscono un peggioramento del glaucoma, pertanto sono necessarie misurazioni periodiche.
6. Fisiopatologia e meccanismo dettagliato di insorgenza
La normale formazione del segmento anteriore segue un complesso programma di sviluppo. All’inizio della terza settimana embrionale, si forma il solco ottico nella placca neurale, segnando l’inizio dello sviluppo dell’organo visivo. Alla fine della terza settimana embrionale si forma la vescicola ottica, e alla quarta settimana si forma la coppa ottica. Intorno alla sesta settimana embrionale inizia la chiusura della fessura embrionale, che si completa alla settima settimana. Il mesenchima che ricopre la superficie anteriore del cristallino si separa, formando la camera anteriore.
Le cellule della cresta neurale si de-epitelializzano dalla cresta neurale e, attraverso una transizione epitelio-mesenchimale, migrano in varie regioni dell’occhio. Le cellule trabecolari derivano dalla cresta neurale, mentre il tessuto connettivo para-canalicolare di Schlemm deriva da cellule endoteliali vascolari; questa differenza di origine forma il principale sito di resistenza al deflusso dell’umore acqueo.
PAX6 : «fattore di controllo master» nello sviluppo dell’occhio. Situato sul cromosoma 11. Coinvolto nell’aniridia, nell’anomalia di Peters e nella sindrome di ectropion dell’iride congenito.
PITX2: fattore di trascrizione. Cromosoma 4 (4q25). Associato sia ai sintomi oculari che uditivi osservati nell’ARS.
FOXC1: fattore di trascrizione. Cromosoma 6 (6p25). Coinvolto nell’ARS, associato sia ai sintomi oculari che a quelli uditivi, come PITX2.
Il glaucoma secondario nell’ASDA si verifica principalmente a causa di una malformazione della via di deflusso dell’umore acqueo. Nello specifico, sono coinvolti i seguenti fattori in modo complesso.
Immaturità del trabecolato: il tessuto connettivo para-canalicolare di Schlemm è anormalmente spesso e la matrice extracellulare si accumula eccessivamente.
Attaccamento del corpo ciliare al trabecolato: la contrazione del muscolo ciliare tira in avanti lo sperone sclerale, comprimendo il canale di Schlemm e il trabecolato.
Inserzione alta della radice dell’iride: la radice dell’iride si trova in corrispondenza del trabecolato, ostacolando il deflusso dell’umore acqueo.
Ipoplasia o assenza del canale di Schlemm.
La sindrome ICE ha un’eziologia diversa rispetto ad altre ASDA. È stata proposta una teoria virale secondo cui il virus dell’herpes simplex (HSV) è coinvolto nella degenerazione delle cellule endoteliali corneali, ma non è stata confermata. È acquisita, si manifesta in adulti di mezza età (leggermente più frequente nelle donne) ed è solitamente unilaterale, differendo anche in questo dalle altre ASDA.
Patologia della cheratopatia associata ad aniridia (AAK)
I pazienti con aniridia sviluppano un progressivo opacamento corneale nel corso della vita. Il meccanismo principale è considerato l’insufficienza delle cellule staminali del limbus corneale (limbal stem cell deficiency; LSCD). Diversi studi che hanno confermato la mutazione PAX6 hanno documentato questo cambiamento progressivo. L’incidenza riportata varia dal 20% a oltre l’80% e si manifesta spesso in modo simmetrico, ma non sempre. 2)
L’analisi dell’esoma e del genoma intero sta portando avanti l’identificazione di nuovi geni correlati. Ancora nel 40-75% dei casi il gene causale non è stato identificato, e l’analisi dei casi irrisolti rimane una sfida importante per il futuro. La chiarificazione della correlazione tra genotipo e fenotipo clinico è attesa per applicazioni nella medicina personalizzata.
Nei casi con mutazioni di FOXC1 e PITX2, l’età di insorgenza e il quadro clinico del glaucoma sono variabili. Sebbene il genotipo possa essere associato alla diversità fenotipica, la stessa mutazione genica può presentare forme cliniche differenti, rendendo difficile la diagnosi e la prognosi. 1)
L’incidenza della cheratopatia nell’aniridia (AAK) è riportata tra il 20 e oltre l’80% e diversi studi con mutazione PAX6 confermata hanno documentato la progressione dell’opacità corneale nel corso della vita. La ricerca sul trapianto di cellule staminali del limbus corneale mirato al LCSD sta progredendo, ma al momento è ancora in fase sperimentale e non è stata stabilita come terapia standard. 2)
Studi epidemiologici hanno mostrato un’associazione tra l’esposizione all’inquinamento atmosferico (PM2.5) prima del concepimento e durante la gravidanza e il rischio di ASDA, e si sta esplorando l’applicazione alla salute pubblica dal punto di vista della medicina preventiva ambientale. Ciò potrebbe portare a future strategie preventive.
Applicazione di dispositivi chirurgici mini-invasivi
L’applicazione di micropulsi laser e dispositivi per la chirurgia del glaucoma minimamente invasiva (MIGS) nei bambini con ASDA è in fase di ricerca. I dati sui risultati a lungo termine sono limitati e l’efficacia e la sicurezza non sono state stabilite come per il glaucoma dell’adulto.
Knight LSW, Ruddle JB, Taranath DA, et al. Childhood and Early Onset Glaucoma Classification and Genetic Profile in a Large Australasian Disease Registry. Ophthalmology. 2021;128(11):1549-1560. doi:10.1016/j.ophtha.2021.04.016.
Hu JCW, Trief D. A narrative review of limbal stem cell deficiency & severe ocular surface disease. Ann Eye Sci. 2023;8:13. doi:10.21037/aes-22-35. https://aes.amegroups.org/article/view/7385/html
Pazos M, Traverso CE, Viswanathan A; European Glaucoma Society. European Glaucoma Society - Terminology and guidelines for glaucoma, 6th Edition. Br J Ophthalmol. 2025;109(Suppl 1):1-212. doi:10.1136/bjophthalmol-2025-egsguidelines. PMID:41026937.