As Anomalias do Desenvolvimento do Segmento Anterior (Anterior Segment Developmental Anomalies; ASDA) são um conjunto de distúrbios do desenvolvimento relacionados ao segmento anterior do olho — córnea, íris, cristalino e câmara anterior. Também são chamadas de Disgenesias do Segmento Anterior (Anterior Segment Dysgenesis; ASD).
As ASDA incluem as seguintes entidades patológicas representativas:
Essas doenças são diversas tanto fenotipicamente quanto geneticamente, e sabe-se que mais de 50 genes estão envolvidos. O conhecimento genético continua a se expandir por meio de sequenciamento de exoma e genoma completo, mas em 40 a 75% dos casos o gene causador ainda não foi identificado. Casos que não podem ser classificados em um fenótipo específico são descritos como “ASD não classificável (unclassified ASD)”. 1)
O humor aquoso produzido pelo corpo ciliar da íris é drenado através da malha trabecular (trabecular meshwork) para o canal de Schlemm (Schlemm’s canal) e também pela via de drenagem uveoescleral. Na ASDA, esse processo é frequentemente prejudicado, e o glaucoma secundário (secondary glaucoma) é uma complicação importante e comum.
Quando apenas o anel embrionário posterior está presente, sem sintomas sistêmicos, ele é tratado de forma distinta da ARS, com base no 9º Relatório de Consenso da Sociedade Mundial de Glaucoma. 1)
QCom que idade as anomalias do desenvolvimento do segmento anterior (ASDA) são diagnosticadas?
A
Depende da doença. O glaucoma congênito primário geralmente se manifesta no primeiro ano de vida. A síndrome de Axenfeld-Rieger e a anomalia de Peters são frequentemente diagnosticadas ao nascimento. O glaucoma de desenvolvimento de início tardio pode ter seu início retardado até a segunda ou terceira década de vida. Em todos os casos, a detecção precoce e o tratamento precoce são importantes.
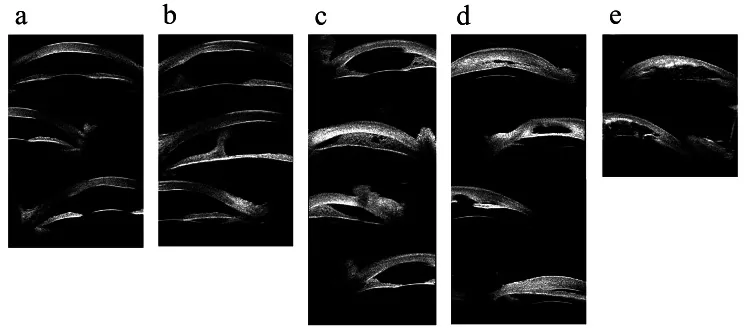
Hong J, et al. Classifications of anterior segment structure of congenital corneal opacity in infants and toddlers by ultrasound biomicroscopy and slit-lamp microscopic photographs: an observational study. BMC Ophthalmol. 2024. Figure 1. PMCID: PMC10804776. License: CC BY.
(a) Opacidade corneana, (b) Opacidade corneana com aderência anterior central, (c) Aderência iridocorneana periférica menor ou igual a 180 graus, (d) Aderência iridocorneana periférica maior que 180 graus, (e) Opacidade corneana com anomalias da íris e do cristalino. Corresponde às aderências do segmento anterior e opacidade corneana discutidas na seção “2. Principais sintomas e achados clínicos”.
Na primeira infância, os seguintes sintomas associados ao aumento da pressão intraocular são frequentemente observados como sintomas iniciais.
Lacrimejamento (epífora): ocorre como resultado da irritação causada pelo edema do epitélio corneano devido ao aumento da pressão intraocular.
Fotofobia (sensibilidade à luz): sintoma que reflete a irritação da córnea.
Blefaroespasmo: surge pelo mesmo mecanismo do lacrimejamento e da fotofobia.
Em crianças mais velhas e adultos, no tipo de início tardio, pode-se notar visão turva ou diminuição da acuidade visual desde idades relativamente jovens. Quando a pressão intraocular está muito alta, podem ocorrer sintomas como fadiga ocular e dor de cabeça. Na aniridia, pode haver queixa de fotofobia (cegueira diurna).
O olho de boi (aumento do diâmetro da córnea) e a opacidade corneana são frequentemente descobertos pelos pais, levando à consulta médica.
A ASDA apresenta achados característicos para cada doença. Os principais achados das entidades nosológicas representativas são mostrados abaixo.
Anel de Schwalbe posterior / ARS
Anel de Schwalbe posterior (PE): Linha de Schwalbe deslocada anteriormente e espessada. Observada como uma linha concêntrica acinzentada na parte interna do limbo corneano à lâmpada de fenda.
Anomalia de Axenfeld: Anel de Schwalbe posterior com aderências em forma de cordão do tecido iridiano periférico.
Anomalia de Rieger: Além do acima, apresenta desvio pupilar, ectrópio de úvea e pseudopolicoria devido à hipoplasia do estroma da íris. Herança autossômica dominante. Glaucoma ocorre em 50-60% dos casos.
Anomalia de Peters
Opacidade corneana central: achado essencial para o diagnóstico. Reflete a ausência do endotélio, membrana de Descemet e estroma corneano.
Tipo 1: apenas ausência da camada posterior da córnea e opacidade corneana.
Tipo 2: com aderência da íris.
Tipo 3: Associado a deslocamento anterior do cristalino e catarata. Cerca de 80% dos casos são bilaterais. Glaucoma está presente em 50-70% dos casos.
Aniridia
Hipoplasia da íris: Predominantemente defeito na porção posterior da íris. Pode estar associada a hipoplasia macular, hipoplasia do nervo óptico e glaucoma.
Ceratopatia associada à aniridia (CAA): A incidência relatada varia de 20 a mais de 80%. É uma opacidade corneana progressiva devido à insuficiência de células-tronco do limbo (ICTL), que progride ao longo da vida. 2)
Síndrome WAGR: ocorre quando há mutação no PAX6 e no gene WT1 adjacente. Inclui tumor de Wilms, aniridia, anomalias geniturinárias e atraso no desenvolvimento mental. 3)
Tipo de anomalia corneana
Megalocórnea: diâmetro da córnea ≥13 mm (≥12 mm em recém-nascidos). Geralmente, a pressão intraocular e a densidade de células endoteliais são normais. Herança ligada ao X recessiva é comum.
Córnea escleralizada: tecido escleral opaco invade a córnea periférica. A borda entre esclera e córnea é indistinta, com invasão vascular.
CHED: Edema corneano bilateral simétrico que aparece ao nascimento ou entre 1-2 anos de idade. Não acompanhado de aumento da pressão intraocular. Herança autossômica recessiva.
Os achados adicionais quando complicado por glaucoma secundário são mostrados abaixo.
Aumento da pressão intraocular: Pode apresentar hipertensão ocular (cerca de 30-50 mmHg).
Aumento do diâmetro corneano (buftalmia): Devido à distensão do revestimento ocular. Se o diâmetro exceder 12,0 mm logo após o nascimento, suspeita-se de glaucoma congênito.
Linhas de Haab: opacidades lineares permanentes que permanecem no local da ruptura da membrana de Descemet.
Escavação aumentada do disco óptico: em lactentes, relação C/D ≥0,3 sugere glaucoma. Diferença entre os olhos ≥0,2 também é um sinal suspeito.
QQual a porcentagem de pacientes com síndrome de Axenfeld-Rieger que desenvolvem glaucoma?
A
O glaucoma se desenvolve em 50-60% (alguns relatos indicam 50-75%) dos casos, sendo uma frequência alta. 3) A herança é autossômica dominante. Casos com manifestações sistêmicas (anomalias dentárias, faciais, hipofisárias, etc.) são denominados síndrome de Rieger. Recomenda-se triagem para glaucoma em familiares.
A principal causa da ASDA são anormalidades genéticas, envolvendo diferentes genes e padrões de herança para cada doença. Os genes causadores das principais doenças são mostrados abaixo.
Além disso, também foram relatados casos de glaucoma de desenvolvimento associados a anormalidades genéticas como PAX6, PITX2 e FOXC1. A correlação entre genótipo e fenótipo é variada, e mesmo dentro de uma família com a mesma anormalidade genética, o fenótipo pode ser diferente.
A maioria dos casos de glaucoma de desenvolvimento de início precoce (glaucoma congênito primário) é esporádica, mas cerca de 10% segue um padrão de herança autossômica recessiva. Também existe a teoria de herança multifatorial.
As células da crista neural (neural crest cells) desempenham um papel central na formação do segmento anterior do olho. As células trabeculares são derivadas da crista neural, enquanto o tecido conjuntivo justacanalicular é derivado de células endoteliais vasculares. O ponto de maior resistência ao fluxo de humor aquoso está na junção desses tecidos de origens diferentes. A síndrome de Axenfeld-Rieger, a anomalia de Peters e a síndrome de ectrópio da íris congênita são consideradas anomalias congênitas resultantes de migração anormal das células da crista neural.
Um grande estudo na Coreia mostrou que o aumento da exposição materna ao PM2,5 (material particulado fino) nos três meses anteriores à concepção e durante o primeiro e segundo trimestres da gravidez foi associado a um maior risco de ASDA na prole.
Exame com lâmpada de fenda: avalia a extensão e localização da opacidade corneana, presença de linhas de Haab, profundidade da câmara anterior, anomalias da íris (embriotoxon posterior, aderências da íris à linha de Schwalbe) e anomalias do cristalino. Verificar a presença de embriotoxon posterior, anomalias da íris (ARS) e catarata (anomalia de Peters).
Medição da pressão intraocular: O tonômetro de aplanação de Goldmann é o padrão, mas para crianças, tonômetros portáteis como o tonômetro de rebote (iCare) ou o tonômetro elétrico (Tonopen) são úteis. Observe que a pressão intraocular diminui sob anestesia geral. Não há intercambialidade dos valores medidos entre diferentes tonômetros.
Medição do diâmetro corneano: Medir os diâmetros horizontal e vertical com um compasso. A faixa normal em recém-nascidos é de 9,5 a 10,5 mm. Se exceder 12,0 mm logo após o nascimento, suspeitar de glaucoma congênito.
Exame do ângulo: Usar uma lâmpada de fenda portátil e um gonioscópio direto, como a lente de Koeppe. Avaliar inserção alta da íris, inserção da íris na linha de Schwalbe (achado ARS) e aumento da largura do trabeculado.
Exame de fundo de olho: Observar a escavação do disco óptico. Em lactentes, uma relação escavação/disco (C/D) ≥ 0,3 sugere glaucoma. A redução da escavação do disco com a diminuição da pressão intraocular é um sinal de bom controle pressórico.
Microscopia ultrassônica de biomicroscopia (UBM): Útil em casos onde a opacidade corneana dificulta a visualização do ângulo. Auxilia na avaliação do grau de anomalia do ângulo e na estimativa do prognóstico da cirurgia de reconstrução da via de drenagem.
OCT de segmento anterior (AS-OCT): Como exame complementar, permite avaliar de forma não invasiva a estrutura do ângulo e da córnea, mas não substitui a gonioscopia no diagnóstico. 3)
Exame de campo visual: Essencial para o diagnóstico de neuropatia óptica glaucomatosa infantil. Em crianças menores de 5 anos, mesmo examinadores experientes encontram dificuldades, sendo a perimetria dinâmica mais fácil de realizar.
De acordo com a 4ª edição das Diretrizes de Prática Clínica para Glaucoma da Sociedade Japonesa de Glaucoma, o diagnóstico de glaucoma infantil é feito quando dois ou mais dos seguintes critérios são atendidos.
Pressão intraocular > 21 mmHg
Progressão do aumento da relação escavação/disco (C/D), assimetria da relação C/D ≥ 0,2, afinamento do anel neuroretiniano
Achados corneanos: linhas de Haab, ou diâmetro corneano ≥ 11 mm em recém-nascidos, ≥ 12 mm em menores de 1 ano, ≥ 13 mm em todas as idades
Progressão da miopia devido ao alongamento do comprimento axial ocular além do desenvolvimento normal
Defeito de campo visual reproduzível consistente com neuropatia óptica glaucomatosa
O diagnóstico diferencial com doenças que apresentam opacidade corneana e aumento do diâmetro corneano é mostrado abaixo.
Megalocórnea: sem aumento da pressão intraocular, escavação do disco óptico ou linhas de Haab. Ângulo normal.
Córnea esclerosada: tecido escleral opaco e invasão vascular.
CHED: edema corneano bilateral simétrico. Sem aumento da pressão intraocular.
Distrofia polimorfa posterior: sem aumento do diâmetro corneano. Exame do endotélio corneano é útil para o diagnóstico.
Trauma de parto com fórceps: opacidade linear unilateral, vertical ou oblíqua.
Doenças metabólicas como mucopolissacaridose congênita e cistinúria: a avaliação dos sintomas sistêmicos é importante para o diagnóstico diferencial.
Os grupos de doenças incluídos na ASD (anomalia de Axenfeld-Rieger, anomalia de Peters, aniridia, distrofia polimorfa posterior, microftalmia, microcórnea, etc.) devem ser considerados mutuamente no diagnóstico diferencial. 3)
QO embriotoxon posterior isolado também está associado ao glaucoma?
A
Casos isolados de embriotoxon posterior (sem sintomas sistêmicos) são diferenciados da ARS, mas também são um dos achados associados à ARS. O embriotoxon posterior pode ser observado em olhos saudáveis e, por si só, não indica necessariamente risco de glaucoma. No entanto, quando associado a outras doenças, como a síndrome de Alagille, é necessário monitorar a pressão intraocular.
Terapia medicamentosa é um tratamento adjuvante com o objetivo de reduzir a pressão intraocular a curto prazo até a cirurgia e controlar a pressão intraocular pós-operatória. A seleção de medicamentos é basicamente a mesma que para glaucoma de ângulo aberto em adultos. No entanto, betabloqueadores requerem cautela quanto a asma brônquica e bradicardia, e há relatos de apneia em neonatos. A acetazolamida oral (5 a 10 mg/kg a cada 6 a 8 horas) também é possível.
O glaucoma de desenvolvimento precoce é basicamente uma doença que requer terapia cirúrgica. A terapia medicamentosa tem um papel adjuvante.
Goniotomia: é adequada como cirurgia inicial em casos com pouca opacidade corneana. Tem a vantagem de não invadir a conjuntiva. Usando uma lente de Barkan ou Swan-Jacob, a superfície da malha trabecular é raspada com uma faca de goniotomia.
Trabeculotomia: pode ser realizada independentemente da presença de opacidade corneana. Também é realizada como cirurgia adicional quando a goniotomia é insuficiente.
Trabeculectomia e cirurgia de tubo de drenagem: opções quando a cirurgia do ângulo é ineficaz. Na ARS, a cirurgia do ângulo é escolhida se o ângulo estiver aberto e a cobertura trabecular por sinéquias anteriores periféricas não for extensa, mas a taxa de sucesso é menor que no PCG. Em casos de falha da cirurgia do ângulo, a trabeculectomia ou a cirurgia de tubo de drenagem com placa podem ser a primeira escolha. 4)
Na anomalia de Peters, o tratamento segue o mesmo padrão do PCG, mas a proporção de casos com boa pressão intraocular pós-operatória é de apenas cerca de 1/3, com muitos casos de mau prognóstico. Devido a anormalidades corneanas associadas, muitas vezes é difícil obter visão útil. 4)
Anomalia de Peters: Em casos leves, a opacidade da córnea frequentemente diminui gradualmente. Se a pressão intraocular for normal, geralmente há alguma melhora e, como o prognóstico após transplante de córnea é ruim, o transplante de córnea na primeira infância geralmente não é realizado. Muitos casos são refratários ao tratamento medicamentoso do glaucoma e, mesmo após cirurgia de reconstrução da via de drenagem, o controle é difícil, com mau prognóstico.
CHED: Para disfunção endotelial da córnea, o transplante de córnea (incluindo transplante endotelial) pode ser indicado.
Córneaesclerocórnea: Pode estar associada a outras síndromes de ASD, e casos graves podem ser candidatos a transplante de córnea.
Fotofobia e fadiga ocular: considerar o uso de óculos com proteção contra luz e lentes de contato cosméticas.
Cerateopatia associada à aniridia (CAA): para opacidade corneana progressiva, considera-se transplante de células-tronco do limbo corneano.
Rastreio de tumor de Wilms: em casos de aniridia esporádica, recomenda-se encaminhamento ao pediatra e realização de rastreio até os 6 anos de idade. 4)
Mesmo com a redução da pressão intraocular, o tratamento da ambliopia é frequentemente necessário. Anisometropia refrativa, astigmatismo irregular, opacidade corneana e linhas de Haab podem causar ambliopia, portanto, exames de acuidade visual e refração devem ser realizados em paralelo com a medição da pressão intraocular. A progressão da miopia e o alongamento axial sugerem progressão do glaucoma, necessitando de medições regulares.
6. Fisiopatologia e mecanismo detalhado de desenvolvimento
A formação normal do segmento anterior segue um programa de desenvolvimento complexo. No início da terceira semana de gestação, o sulco óptico se forma na placa neural, marcando o início do desenvolvimento do órgão visual. No final da terceira semana, a vesícula óptica se forma, e na quarta semana, o cálice óptico se desenvolve. Por volta da sexta semana, a fissura embrionária começa a se fechar, completando-se na sétima semana. O mesênquima que cobre a superfície anterior do cristalino se separa para formar a câmara anterior.
As células da crista neural se desepitelizam da crista neural, passam por uma transição epitélio-mesenquimal e migram para várias regiões dentro do olho. As células trabeculares são derivadas da crista neural, enquanto o tecido conjuntivo justacanalicular é derivado de células endoteliais vasculares; essa diferença de origem forma o local de maior resistência ao fluxo do humor aquoso.
PAX6: «Fator mestre de controle» no desenvolvimento ocular. Localizado no cromossomo 11. Envolvido na aniridia, anomalia de Peters e síndrome de ectrópio da íris congênita.
PITX2: Fator de transcrição. Cromossomo 4 (4q25). Associado tanto aos sintomas oculares quanto auditivos observados na ARS.
FOXC1: Fator de transcrição. Cromossomo 6 (6p25). Envolvido na ARS, associado tanto aos sintomas oculares quanto auditivos, similarmente ao PITX2.
O glaucoma secundário na ASDA ocorre principalmente devido à malformação da via de drenagem do humor aquoso. Especificamente, os seguintes fatores estão envolvidos de forma combinada.
Imaturidade do desenvolvimento da malha trabecular: o tecido conjuntivo justacanalicular é anormalmente espesso, com acúmulo excessivo de matriz extracelular.
Inserção do corpo ciliar na malha trabecular: a contração do músculo ciliar puxa o esporão escleral para frente, comprimindo o canal de Schlemm e a malha trabecular.
Inserção alta da raiz da íris: a raiz da íris está localizada na posição da malha trabecular, obstruindo o fluxo do humor aquoso.
Hipoplasia ou ausência do canal de Schlemm.
A síndrome ICE difere de outras ASDA em sua etiologia. A teoria viral, envolvendo o vírus herpes simplex (HSV) na degeneração das células endoteliais da córnea, foi proposta, mas não confirmada. É adquirida, ocorre em adultos de meia-idade (ligeiramente mais comum em mulheres) e geralmente é unilateral, diferindo também de outras ASDA.
Patogênese da ceratopatia associada à aniridia (AAK)
Pacientes com aniridia desenvolvem opacidade progressiva da córnea ao longo da vida. A insuficiência de células-tronco do limbo (LSCD) é considerada o principal mecanismo. Múltiplos estudos com mutações confirmadas no PAX6 documentaram essa alteração progressiva. A incidência relatada varia de 20 a mais de 80%, geralmente aparecendo simetricamente, mas nem sempre. 2)
A análise de exoma e genoma completo está avançando na identificação de novos genes relacionados. Ainda em 40 a 75% dos casos o gene causador não é identificado, e a análise dos casos não resolvidos remanescentes é uma questão importante para o futuro. Espera-se que o esclarecimento da correlação entre genótipo e fenótipo clínico contribua para a aplicação na medicina personalizada.
Em casos com mutações em FOXC1 e PITX2, há variação na idade de início e nas características clínicas do glaucoma. Embora o genótipo possa estar associado à diversidade fenotípica, o fato de que a mesma mutação genética pode resultar em diferentes formas da doença torna o diagnóstico e a previsão do prognóstico desafiadores. 1)
A incidência de ceratopatia na aniridia (AAK) é relatada entre 20% e mais de 80%, e múltiplos estudos com mutação PAX6 confirmada documentaram a progressão da opacidade corneana ao longo da vida. A pesquisa sobre transplante de células-tronco do limbo corneano direcionado ao LCSD está avançando, mas, no momento, ainda está em fase de pesquisa e não foi estabelecida como tratamento padrão. 2)
Estudos epidemiológicos mostram associação entre exposição à poluição do ar (PM2.5) antes e durante a gravidez e o risco de ASDA, e a aplicação em saúde pública está sendo explorada sob a perspectiva da medicina preventiva ambiental. Isso pode levar a futuras estratégias de prevenção.
Aplicação de dispositivos cirúrgicos minimamente invasivos
A aplicação de laser de micropulso e dispositivos de cirurgia minimamente invasiva do glaucoma (MIGS) em crianças com ASDA está em fase de pesquisa. Os dados de resultados de longo prazo são limitados, e a eficácia e segurança equivalentes às do glaucoma em adultos não foram estabelecidas.
Knight LSW, Ruddle JB, Taranath DA, et al. Childhood and Early Onset Glaucoma Classification and Genetic Profile in a Large Australasian Disease Registry. Ophthalmology. 2021;128(11):1549-1560. doi:10.1016/j.ophtha.2021.04.016.
Hu JCW, Trief D. A narrative review of limbal stem cell deficiency & severe ocular surface disease. Ann Eye Sci. 2023;8:13. doi:10.21037/aes-22-35. https://aes.amegroups.org/article/view/7385/html
Pazos M, Traverso CE, Viswanathan A; European Glaucoma Society. European Glaucoma Society - Terminology and guidelines for glaucoma, 6th Edition. Br J Ophthalmol. 2025;109(Suppl 1):1-212. doi:10.1136/bjophthalmol-2025-egsguidelines. PMID:41026937.