前眼部發育異常(ASDA)是影響角膜 、虹膜 、水晶體 等前眼部結構形成的先天性疾病的統稱。
涉及50多個基因,其表現型和基因型均具有異質性特徵。
房水 流出通道發育不全導致的續發性青光眼 是常見的主要併發症,需要早期進行眼壓 管理。根據表現型不同,存在不同的疾病實體,如後胚胎環、Axenfeld-Rieger症候群 、Peters異常 和無虹膜 症。
合併青光眼 時,除藥物治療外,隅角 手術(隅角 切開術或小樑切開術 )是基本治療。
神經嵴細胞的遷移和分化異常是許多疾病的共同胚胎學背景。
仍有40%–75%的病例致病基因不明,需要持續的遺傳學研究。
前眼部發育異常(Anterior Segment Developmental Anomalies; ASDA)是與眼前部——角膜 、虹膜 、水晶體 、前房 ——相關的發育障礙的總稱。也稱為前眼部形成異常(Anterior Segment Dysgenesis; ASD )。
ASDA包括以下代表性疾病。
這些疾病在表現型和基因型上都具有多樣性,已發現超過50個基因與之相關。儘管透過外顯子組分析和全基因組分析,遺傳學知識不斷擴展,但仍有40%–75%的病例未能確定致病基因。無法歸類為特定表現型的病例被描述為「未分類ASD 」。1)
由虹膜 睫狀體 產生的房水 通過小樑網進入施萊姆管,並經由葡萄膜鞏膜 流出通路排出。在ASDA中,此過程常受損,繼發性青光眼 是常見且重要的併發症。
如果僅存在後胚胎環而無全身症狀,則根據世界青光眼 協會第9次共識報告,將其與ARS區分處理。1)
Q
前眼部發育異常(ASDA)通常在幾歲時被診斷?
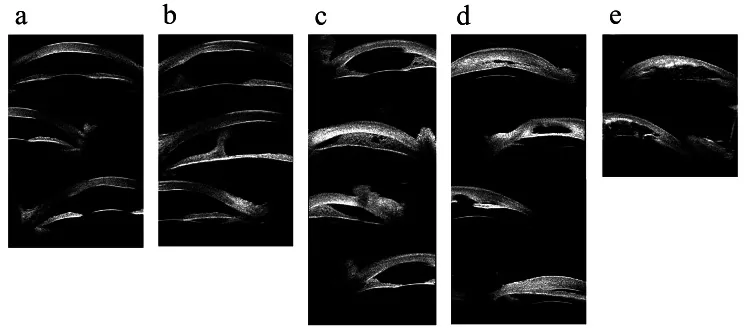
Hong J, et al. Classifications of anterior segment structure of congenital corneal opacity in infants and toddlers by ultrasound biomicroscopy and slit-lamp microscopic photographs: an observational study. BMC Ophthalmol. 2024. Figure 1. PMCID: PMC10804776. License: CC BY.
(a)
角膜 混濁、(b)
角膜 混濁伴中央前粘連、(c)≤180度的周邊
虹膜 角膜 粘連、(d)>180度的周邊
虹膜 角膜 粘連、(e)伴有
虹膜 和晶狀體異常的
角膜 混濁的
超音波生物顯微鏡 圖像。這些對應於「2. 主要症狀和臨床所見」一節中討論的
前房 粘連和
角膜 混濁。
在嬰幼兒期,眼壓 升高引起的以下症狀常作為首發症狀出現。
流淚(淚液過多) :由於眼壓 升高導致角膜上皮 水腫,刺激引起。畏光 (光敏感)角膜 刺激的症狀。眼瞼痙攣 畏光 機制相同。
在年齡較大的兒童和成人中,遲發型病例可能從相對年輕時就開始出現視物模糊或視力 下降。當眼壓 非常高時,可能出現眼疲勞 或頭痛等症狀。無虹膜 症患者可能主訴畏光 (晝盲)。
牛眼(角膜 直徑增大)和角膜 混濁常由家長發現,從而促使就醫。
ASDA根據疾病不同呈現特徵性表現。以下列出代表性疾病單元的主要所見。
後胚胎環 / ARS
後胚胎環(PE) :向前移位並增厚的Schwalbe線。在裂隙燈 顯微鏡下,角膜緣 內側可見灰白色同心圓線。
阿克森費爾德異常 :後胚胎環伴有周邊虹膜 組織索狀黏連。
里格異常 :除上述表現外,因虹膜 基質發育不全導致瞳孔 偏位、葡萄膜外翻和假性多瞳。體染色體顯性遺傳 。50%~60%併發青光眼 。
彼得斯異常
角膜 中央混濁角膜內皮 、後彈力層和角膜基質 的缺損 。
1型 :僅角膜 後部缺損 和角膜 混濁。
2型 :伴有虹膜 黏連。
第3型 :伴有水晶體 前移及白內障 。約80%為雙眼性。50~70%合併青光眼 。
無虹膜症
虹膜 發育不全虹膜 後部缺損 。可能伴有黃斑部 發育不全、視神經發育不全 及青光眼 。
無虹膜 相關角膜 病變(AAK) :發生率報告為20%~80%以上。由角膜緣幹細胞 缺乏(LSCD )引起的進行性角膜 混濁,終生進展。2)
WAGR症候群 :由PAX6與相鄰的WT1基因突變引起。包括威爾姆氏腫瘤、無虹膜 、泌尿生殖系統異常及智能障礙。3)
角膜異常型
巨大角膜 :角膜 直徑≥13mm(新生兒≥12mm)。通常眼壓 和內皮細胞密度正常。多為X染色體隱性遺傳。
鞏膜化角膜 鞏膜 組織侵入周邊角膜 。鞏膜 與角膜 邊界不清,伴有血管侵入。
CHED 角膜水腫 。不伴有眼壓 升高。體染色體隱性遺傳 。
合併續發性青光眼 時,可能出現以下表現。
眼壓 升高眼壓 (約30-50 mmHg)。角膜 直徑增大(牛眼)先天性青光眼 。Haab線 :Descemet膜破裂處殘留的永久性線狀混濁。視神經 盤凹陷擴大青光眼 。雙眼差異≥0.2也是可疑徵象。
Q
Axenfeld-Rieger症候群患者中,青光眼的發生率是多少?
A
50–60%(部分報告為50–75%)的患者發生青光眼 ,頻率很高。3) 呈體染色體顯性遺傳 。伴有全身症狀(牙齒異常、顏面骨異常、垂體異常等)者稱為Rieger症候群。建議對親屬進行青光眼篩檢 。
ASDA的主要原因是遺傳異常,不同疾病涉及不同的基因和遺傳模式。主要疾病的致病基因如下所示。
疾病 主要致病基因 遺傳模式 ARS PITX2(4q25)、FOXC1(6p25) 體染色體顯性遺傳 彼得斯異常 PAX6、PITX2、CYP1B1 散發性、顯性、隱性 原發性先天性青光眼 CYP1B1(GLC3A)、LTBP2(GLC3C) 體染色體隱性遺傳 無虹膜 症 PAX6(11號染色體) 體染色體顯性 CHED SLC4A11、ZEB1 體染色體隱性 巨大角膜 CHRDL1 X染色體隱性遺傳
此外,也報告了伴有PAX6、PITX2、FOXC1等基因異常的發育性青光眼 。基因型與表現型的關聯多樣,即使在同一基因異常的家族內,表現型也可能不同。
早發型發育性青光眼 (原發性先天性青光眼 )大部分為散發病例,但約10%為體染色體隱性遺傳 。也有多因子遺傳的說法。
神經嵴細胞 在前眼部形成中扮演核心角色。小梁網 細胞源自神經嵴,而鄰近許萊姆管 結締組織源自血管內皮細胞。這些起源不同的組織相鄰處存在最大的房水 流出阻力。ARS、彼得斯異常 和先天性虹膜 外翻症候群均被視為由神經嵴細胞遷移異常引起的先天異常。
韓國的一項大規模研究顯示,母親在受孕前三個月以及妊娠第一和第二孕期暴露於PM2.5(細懸浮微粒)增加,與後代ASDA風險升高相關。
ASDA的診斷主要基於臨床。5歲及以下兒童進行檢查時,通常需要在全身麻醉或鎮靜下進行。
裂隙燈顯微鏡檢查 角膜 混濁的程度和部位、Haab線的存在、前房 深度、虹膜 異常(後胚胎環、虹膜 附著於Schwalbe線)以及水晶體 異常。檢查後胚胎環、虹膜 異常(ARS)和白內障 (Peter異常)。眼壓測量 Goldmann壓平眼壓計 是標準,但兒童可使用攜帶式眼壓 計,如回彈式眼壓 計(iCare)或電子眼壓 計(Tono-Pen)。注意全身麻醉下眼壓 會降低。不同眼壓 計之間的測量值不可互換。角膜 直徑測量先天性青光眼 。隅角檢查 裂隙燈 和直接隅角 鏡如Koeppe鏡。評估虹膜 高位插入、虹膜 附著於Schwalbe線(ARS表現)以及小梁網 寬度增加。眼底檢查 視神經 盤凹陷。嬰幼兒杯盤比≥0.3時懷疑青光眼 。眼壓 下降後凹陷縮小表示眼壓 控制良好。超音波生物顯微鏡 (UBM )角膜 混濁導致隅角 難以觀察的病例。有助於評估隅角 發育異常程度及預測流出道重建手術的預後。前眼部OCT (AS-OCT )隅角 和角膜 結構,但不能取代隅角鏡檢查 進行診斷。3) 視野檢查 青光眼 性視神經病變 所必需。5歲以下兒童即使由熟練檢查者操作也很困難,動態視野檢查 較易實施。
根據日本青光眼 學會青光眼 診療指引第4版,滿足以下2項或以上即可診斷為兒童青光眼 。
眼壓 > 21 mmHg杯盤比進行性增大、杯盤比左右不對稱 ≥ 0.2、視盤邊緣變薄
角膜 表現:Haab線,或新生兒角膜 直徑 ≥ 11 mm,1歲以下 ≥ 12 mm,所有年齡 ≥ 13 mm眼軸長度 超過正常發育的伸長導致近視 進展與青光眼 性視神經病變 一致的可重複性視野缺損
與表現為角膜 混濁和角膜 直徑增大的疾病的鑑別診斷如下所示。
巨大角膜 :無眼壓 升高、視盤凹陷 擴大、Haab線。隅角 正常。鞏膜化角膜 鞏膜 組織伴血管侵入。CHED 角膜水腫 。無眼壓 升高。後部多形性角膜 變性症 :無角膜 直徑增大。角膜內皮 檢查有助於診斷。產鉗分娩外傷 :單眼、垂直或斜行線狀混濁。先天性代謝疾病,如黏多醣症和胱胺酸尿症 :全身症狀的評估對鑑別診斷很重要。
ASD 包含的疾病群(Axenfeld-Rieger異常、Peters異常 、無虹膜 症、後部多形性失養症、小眼球、小角膜 等)需要在鑑別診斷中相互考慮。3)
Q
單獨的後胚胎環(posterior embryotoxon)是否也會合併青光眼?
A
單獨的後胚胎環(無全身症狀)與ARS不同,但也是ARS的合併表現之一。後胚胎環也可能在健康眼中觀察到,其本身不一定意味著青光眼 風險。但若伴有其他疾病如Alagille症候群,則需要監測眼壓 。
ASDA相關青光眼 的治療參照原發性先天性青光眼 (PCG)的治療。
藥物治療 是旨在術前短期降眼壓 和術後眼壓 控制的輔助治療。藥物選擇基本與成人開角型青光眼 相同。但β受體阻滯劑 需注意支氣管哮喘和心動過緩,新生兒有呼吸暫停的報告。也可口服乙醯唑胺 (5~10 mg/kg,每6~8小時一次)。
早發型發育性青光眼 基本需要手術治療 。藥物治療為輔助地位。
隅角 切開術(goniotomy)角膜 混濁較輕的病例的初次手術。優點是不損傷結膜 。使用Barkan鏡或Swan-Jacob鏡,用隅角 切開刀刮除隅角 小梁網 表面。小樑切開術 (trabeculotomy)角膜 混濁與否均可施行。也作為隅角 切開術效果不佳時的追加手術。小樑切除術 /管分流手術隅角 手術無效時的選擇。在ARS中,若隅角 開放且周邊虹膜 前沾黏覆蓋小樑網的範圍不廣,則選擇隅角 手術,但成功率低於PCG。隅角 手術無效時,小樑切除術 或帶板管分流手術可作為首選。4)
彼得斯異常 的治療參照PCG,但術後獲得良好眼壓 的比例僅約手術病例的1/3,預後不良者多。由於伴有角膜 異常等,常難以獲得實用視力 。4)
無虹膜 症伴隨的青光眼 也參照PCG進行治療。4)
彼得斯異常 角膜 混濁常逐漸減輕。若眼壓 正常,多有改善,且因角膜移植術 後預後不良,通常不在幼兒期進行角膜移植 。對青光眼 藥物治療抵抗,即使進行流出道重建手術也難以控制的預後不良病例較多。
CHED 角膜內皮 功能不全,可考慮角膜移植 (包括內皮移植)。
鞏膜化角膜 ASD 症候群相關,重症病例可考慮角膜移植 。
畏光 與眼睛疲勞無虹膜 相關角膜 病變(AAK) :對於進行性角膜 混濁,可考慮角膜緣幹細胞 移植。Wilms腫瘤篩檢 :對於散發性無虹膜 症,建議轉診至兒科並在6歲前進行篩檢。4)
即使眼壓 下降,弱視 治療也常常是必要的。由於屈光參差 、不規則散光 、角膜 混濁和Haab線等可導致弱視 ,視力 及屈光 檢查應與眼壓測量 並行持續進行。近視 進展和眼軸 延長提示青光眼 進展,因此需要定期測量。
正常前節形成遵循複雜的發育程序。妊娠第3週初,神經板形成視溝,是視覺器官發育的開始。第3週末形成視泡,第4週形成視杯。第6週左右開始閉合胚裂,第7週完成。覆蓋水晶體 前面的間充質分離形成前房 。
神經嵴細胞從神經嵴脫上皮化,經過上皮-間質轉化遷移到眼內各處。小樑細胞來源於神經嵴,近施萊姆管結締組織來源於血管內皮細胞,這種起源差異形成了房水 流出最大阻力的部位。
PAX6 :眼睛發育的「主控因子」。位於11號染色體。與無虹膜 症、彼得斯異常 、先天性虹膜 外翻症候群相關。PITX2 :轉錄因子。位於4號染色體(4q25)。與Axenfeld-Rieger症候群 的眼部和聽覺症狀均相關。FOXC1 :轉錄因子。位於6號染色體(6p25)。參與Axenfeld-Rieger症候群 ,與PITX2類似,與眼部和聽覺症狀均相關。CYP1B1 :細胞色素P450家族酶(GLC3A位點)。與原發性先天性青光眼 、彼得斯異常 、鞏膜化角膜 等相關。CHRDL1 :與角膜基質 和內皮發育相關。X染色體連鎖巨大角膜 的致病基因。B3GLCT :彼得斯加氏症候群的致病基因。與糖基化缺陷相關。體染色體隱性遺傳 。
ASDA中的續發性青光眼 主要由於房水 流出通道發育不全引起。具體涉及以下複合因素。
小樑網發育不成熟:近Schlemm管結締組織異常增厚,細胞外基質過度積聚。
睫狀體 附著於小樑網部位:睫狀肌收縮將鞏膜 突向前拉,壓迫Schlemm管和小樑網。虹膜 根部高位附著:虹膜 根部位於小樑網位置,阻礙房水 流出。Schlemm管發育不良或缺失。
ICE症候群 的病因與其他ASDA不同。有病毒病因學說認為單純疱疹病毒(HSV)參與角膜內皮 細胞變性,但尚未確定。該病為後天性,發生於中年成人(女性略多),通常為單眼,這也與其他ASDA不同。
無虹膜 患者終生出現進行性角膜 混濁。角膜緣幹細胞 缺乏(LSCD )被認為是主要機制。多項確認PAX6突變的研究記錄了這種進行性變化。發生率報導為20-80%以上,常對稱出現但不總是如此。2)
透過外顯子組分析和全基因組分析,新的相關基因的鑑定正在推進。然而,仍有40-75%的病例未能確定致病基因,對剩餘的「未解病例」進行分析是未來的重要課題。闡明基因型與臨床表型之間的關聯有望應用於個人化醫療。
在帶有FOXC1和PITX2突變的病例中,青光眼 的發病年齡和臨床表現存在差異。雖然基因型可能與表型多樣性相關,但同一基因突變也可能呈現不同的疾病類型,這使得診斷和預後預測變得困難。1)
無虹膜 症相關角膜 病變(AAK)的發生率據報導為20%至80%以上,多項經PAX6突變確認的研究記錄了終生角膜 混濁的進展。針對LCSD的角膜緣幹細胞 移植研究正在進展中,但目前仍處於研究階段,尚未確立為標準治療。2)
流行病學研究顯示,受精前及懷孕期間暴露於大氣污染(PM2.5)與ASDA風險相關,從環境預防醫學的角度正在探索公共衛生應用。這可能為未來的預防策略提供方向。
微脈衝雷射 和微創青光眼手術 (MIGS )設備在ASDA兒童中的應用處於研究階段。長期結果數據有限,尚未確立與成人青光眼 同等的有效性和安全性。
Knight LSW, Ruddle JB, Taranath DA, et al. Childhood and Early Onset Glaucoma Classification and Genetic Profile in a Large Australasian Disease Registry. Ophthalmology. 2021;128(11):1549-1560. doi:10.1016/j.ophtha.2021.04.016.
Hu JCW, Trief D. A narrative review of limbal stem cell deficiency & severe ocular surface disease. Ann Eye Sci. 2023;8:13. doi:10.21037/aes-22-35. https://aes.amegroups.org/article/view/7385/html
Pazos M, Traverso CE, Viswanathan A; European Glaucoma Society. European Glaucoma Society - Terminology and guidelines for glaucoma, 6th Edition. Br J Ophthalmol. 2025;109(Suppl 1):1-212. doi:10.1136/bjophthalmol-2025-egsguidelines. PMID:41026937.
日本緑内障学会緑内障診療ガイドライン改訂委員会. 緑内障診療ガイドライン(第5版). 日眼会誌. 2022;126(2):85-177.