Les anomalies du développement du segment antérieur (Anterior Segment Developmental Anomalies; ASDA) sont un terme générique désignant les troubles du développement affectant le segment antérieur de l’œil — la cornée, l’iris, le cristallin et la chambre antérieure. On les appelle également « dysgénésie du segment antérieur (Anterior Segment Dysgenesis; ASD) ».
L’ASDA comprend les entités pathologiques représentatives suivantes.
Ces maladies sont diverses tant sur le plan phénotypique que génotypique, et plus de 50 gènes sont impliqués. Les connaissances génétiques continuent de s’élargir grâce au séquençage de l’exome et du génome entier, mais la cause génétique reste non identifiée dans 40 à 75 % des cas. Les cas qui ne peuvent être classés dans un phénotype spécifique sont décrits comme « ASD non classifié ». 1)
L’humeur aqueuse produite par le corps ciliaire de l’iris est évacuée à travers le trabéculum (trabecular meshwork) vers le canal de Schlemm, ainsi que par la voie uvéosclérale. Dans l’ASDA, ce processus est souvent perturbé, faisant du glaucome secondaire une complication fréquente et importante.
Lorsque seul l’anneau embryonnaire postérieur est présent sans symptômes systémiques, il est traité séparément de l’ARS conformément au 9e rapport de consensus de la Société mondiale du glaucome. 1)
QÀ quel âge l'anomalie du développement du segment antérieur (ASDA) est-elle généralement diagnostiquée ?
A
Cela varie selon la maladie. Le glaucome congénital primitif survient souvent dans la première année de vie. Le syndrome d’Axenfeld-Rieger et l’anomalie de Peters sont souvent diagnostiqués dès la naissance. Le glaucome développemental tardif peut survenir jusqu’à l’âge de 10 à 20 ans. Dans tous les cas, un diagnostic précoce et un traitement précoce sont importants.
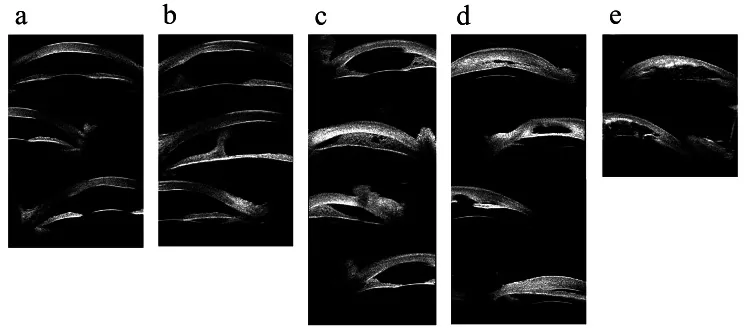
Hong J, et al. Classifications of anterior segment structure of congenital corneal opacity in infants and toddlers by ultrasound biomicroscopy and slit-lamp microscopic photographs: an observational study. BMC Ophthalmol. 2024. Figure 1. PMCID: PMC10804776. License: CC BY.
Il s’agit d’images de biomicroscopie ultrasonore de (a) opacité cornéenne, (b) opacité cornéenne avec synéchie antérieure centrale, (c) synéchie iridocornéenne périphérique de 180 degrés ou moins, (d) synéchie iridocornéenne périphérique de plus de 180 degrés, et (e) opacité cornéenne avec anomalies de l’iris et du cristallin. Elles correspondent aux synéchies du segment antérieur et aux opacités cornéennes traitées dans la section « 2. Principaux symptômes et signes cliniques ».
Dans la petite enfance, les symptômes suivants associés à une augmentation de la pression intraoculaire sont souvent observés comme premiers signes.
Larmoiement (épiphora) : résulte de l’irritation due à l’œdème de l’épithélium cornéen provoqué par l’augmentation de la pression intraoculaire.
Photophobie (sensibilité à la lumière) : symptôme reflétant une irritation cornéenne.
Blepharospasme : apparaît par le même mécanisme que le larmoiement et la photophobie.
Chez les enfants plus âgés et les adultes, dans les formes tardives, une vision floue ou une baisse de l’acuité visuelle peuvent être ressenties dès un âge relativement jeune. En cas de pression intraoculaire très élevée, des symptômes tels que la fatigue oculaire ou des maux de tête peuvent apparaître. Dans l’aniridie, une photophobie (cécité diurne) peut être rapportée.
La buphtalmie (augmentation du diamètre cornéen) et l’opacité cornéenne sont souvent découvertes par les parents, ce qui motive la consultation.
L’ASDA présente des signes caractéristiques selon la maladie. Les principaux signes des entités pathologiques représentatives sont indiqués ci-dessous.
Anneau de Schwalbe postérieur / ARS
Anneau embryotoxique postérieur (PE) : ligne de Schwalbe déplacée vers l’avant et épaissie. Observée à la lampe à fente comme une ligne concentrique gris-blanc à l’intérieur du limbe cornéen.
Anomalie d’Axenfeld : embryotoxon postérieur avec adhérences en bride du tissu irien périphérique.
Anomalie de Rieger : en plus de ce qui précède, présente une déviation pupillaire, une ectropion uvéal et une pseudopolycorie dus à une hypoplasie du stroma irien. Transmission autosomique dominante. Un glaucome survient dans 50 à 60 % des cas.
Anomalie de Peters
Opacité cornéenne centrale : signe essentiel au diagnostic. Elle reflète une perte de l’endothélium cornéen, de la membrane de Descemet et du stroma cornéen.
Type 1 : défaut de la face postérieure de la cornée et opacité cornéenne uniquement.
Type 3 : associé à un déplacement antérieur du cristallin ou à une cataracte. Environ 80 % des cas sont bilatéraux. Un glaucome est présent dans 50 à 70 % des cas.
Aniridie
Hypoplasie de l’iris : déficit principalement de la partie postérieure de l’iris. Peut être associée à une hypoplasie maculaire, une hypoplasie du nerf optique ou un glaucome.
Kératopathie associée à l’aniridie (AAK) : l’incidence rapportée varie de 20 à plus de 80 %. Il s’agit d’une opacité cornéenne progressive due à une insuffisance de cellules souches du limbe (LSCD), qui progresse tout au long de la vie. 2)
Syndrome WAGR : survient lorsque les gènes PAX6 et WT1 adjacent sont mutés. Comprend la tumeur de Wilms, l’aniridie, les anomalies urogénitales et le retard du développement psychomoteur. 3)
Type d'anomalie cornéenne
Mégalocornée : diamètre cornéen ≥13 mm (≥12 mm chez le nouveau-né). La pression intraoculaire et la densité des cellules endothéliales sont généralement normales. Transmission récessive liée à l’X fréquente.
Sclérose cornéenne : infiltration du tissu scléral opaque dans la cornée périphérique. La limite entre la sclère et la cornée est floue, avec une invasion vasculaire.
CHED : Œdème cornéen bilatéral symétrique apparaissant de la naissance à l’âge de 1-2 ans. Sans augmentation de la pression intraoculaire. Autosomique récessif.
Les signes suivants s’ajoutent en cas de glaucome secondaire.
Élévation de la pression intraoculaire : Une pression intraoculaire élevée (environ 30 à 50 mmHg) peut être observée.
Augmentation du diamètre cornéen (œil de bœuf) : due à l’étirement de la membrane oculaire. Un diamètre supérieur à 12,0 mm juste après la naissance fait suspecter un glaucome congénital.
Ligne de Haab : opacité linéaire permanente résiduelle au site de rupture de la membrane de Descemet.
Élargissement de l’excavation papillaire : chez les nourrissons, un rapport C/D ≥ 0,3 fait suspecter un glaucome. Une différence de 0,2 ou plus entre les deux yeux est également un signe évocateur.
QQuel pourcentage de patients atteints du syndrome d'Axenfeld-Rieger développe un glaucome ?
A
On estime que 50 à 60 % (certains rapports indiquent 50 à 75 %) des patients développent un glaucome, ce qui est fréquent. 3) La transmission est autosomique dominante. Les cas présentant des symptômes systémiques (anomalies dentaires, anomalies des os du visage, anomalies hypophysaires, etc.) sont appelés syndrome de Rieger. Un dépistage du glaucome chez les membres de la famille est recommandé.
La cause principale de l’ASDA est une anomalie génétique, impliquant différents gènes et modes de transmission selon la maladie. Les gènes responsables des principales maladies sont présentés ci-dessous.
D’autres glaucomes développementaux associés à des anomalies génétiques telles que PAX6, PITX2, FOXC1 ont également été rapportés. La corrélation entre génotype et phénotype est variable, et même au sein d’une famille présentant la même anomalie génétique, le phénotype peut différer.
La plupart des cas de glaucome développemental précoce (glaucome congénital primaire) sont sporadiques, mais environ 10 % suivent un mode de transmission autosomique récessif. Une hérédité multifactorielle est également évoquée.
Les cellules de la crête neurale (neural crest cells) jouent un rôle central dans la formation du segment antérieur de l’œil. Les cellules trabéculaires sont dérivées de la crête neurale, tandis que le tissu conjonctif juxtacanaliculaire est dérivé des cellules endothéliales vasculaires. La plus grande résistance à l’écoulement de l’humeur aqueuse se situe au point de contact entre ces tissus d’origines différentes. L’ARS, l’anomalie de Peters et le syndrome d’ectropion uvéal congénital sont tous considérés comme des anomalies congénitales résultant d’une migration anormale des cellules de la crête neurale.
Une étude à grande échelle en Corée a montré qu’une exposition accrue des mères aux PM2,5 (particules fines) au cours des trois mois précédant la conception et des premier et deuxième trimestres de grossesse était associée à un risque accru de DAAS chez l’enfant.
Le diagnostic de l’ASDA est principalement clinique. Chez les enfants de moins de 5 ans, les examens nécessitent souvent une anesthésie générale ou une sédation.
Examen à la lampe à fente : évaluer le degré et la localisation de l’opacité cornéenne, la présence de la ligne de Haab, la profondeur de la chambre antérieure, les anomalies iriennes (anneau embryotoxique postérieur, adhérences iriennes à la ligne de Schwalbe) et les anomalies du cristallin. Vérifier la présence d’un anneau embryotoxique postérieur, d’anomalies iriennes (ARS) et de cataracte (anomalie de Peters).
Mesure de la pression intraoculaire : Le tonomètre à aplanation de Goldmann est la référence, mais chez l’enfant, les tonomètres portables comme le tonomètre à rebond (iCare) ou le tonomètre électronique (Tono-Pen) sont utiles. Noter que la pression intraoculaire diminue sous anesthésie générale. Les mesures ne sont pas interchangeables entre différents types de tonomètres.
Mesure du diamètre cornéen : mesure du diamètre horizontal et vertical à l’aide d’un compas. La plage normale chez le nouveau-né est de 9,5 à 10,5 mm. Si le diamètre dépasse 12,0 mm juste après la naissance, suspecter un glaucome congénital.
Examen de l’angle : Utilisation d’une lampe à fiche portative et d’un gonioscope direct tel que la lentille de Koeppe. Évaluer l’insertion haute de l’iris, l’adhérence de l’iris à la ligne de Schwalbe (signe ARS) et l’augmentation de la largeur du trabéculum.
Examen du fond d’œil : observation de l’excavation de la papille optique. Chez les nourrissons, un rapport C/D ≥ 0,3 fait suspecter un glaucome. Une réduction de l’excavation papillaire après baisse de la pression intraoculaire est un signe de bon contrôle de la pression.
Microscope ultrasonique biomicroscopique (UBM) : utile dans les cas où l’opacité cornéenne rend difficile la visualisation de l’angle. Aide également à évaluer le degré de dysgénésie de l’angle et à estimer le pronostic après une chirurgie de reconstruction de la voie d’écoulement.
OCT du segment antérieur (AS-OCT) : en tant qu’examen complémentaire, il permet d’évaluer de manière non invasive la structure de l’angle et de la cornée, mais ne remplace pas la gonioscopie pour le diagnostic. 3)
Examen du champ visuel : indispensable au diagnostic de neuropathie optique glaucomateuse chez l’enfant. Chez les enfants de moins de 5 ans, il est difficile même pour un examinateur expérimenté, et l’examen dynamique du champ visuel est plus facile à réaliser.
Selon la 4e édition des directives de pratique clinique pour le glaucome de la Société japonaise du glaucome, un glaucome pédiatrique est diagnostiqué lorsque deux ou plusieurs des critères suivants sont remplis.
Pression intraoculaire > 21 mmHg
Progression de l’augmentation du rapport C/D, asymétrie du rapport C/D ≥ 0,2, amincissement du rebord
Signes cornéens : stries de Haab, ou chez le nouveau-né diamètre cornéen ≥ 11 mm, chez l’enfant de moins d’un an ≥ 12 mm, à tout âge ≥ 13 mm
Progression de la myopie due à un allongement dépassant le développement normal de la longueur axiale de l’œil
Déficit visuel reproductible correspondant à une neuropathie optique glaucomateuse
Le diagnostic différentiel avec les maladies présentant une opacité cornéenne et une augmentation du diamètre cornéen est présenté ci-dessous.
Mégalocornée : pas d’augmentation de la pression intraoculaire, pas d’élargissement de l’excavation papillaire, pas de lignes de Haab. Angle iridocornéen normal.
Sclérose cornéenne : tissu scléral opaque avec invasion vasculaire.
CHED : œdème cornéen bilatéral symétrique. Pas d’augmentation de la pression intraoculaire.
Dystrophie endothéliale postérieure polymorphe : pas d’augmentation du diamètre cornéen. L’examen de l’endothélium cornéen est utile au diagnostic.
Traumatisme obstétrical par forceps : opacité linéaire unilatérale, à orientation verticale ou oblique.
Maladies métaboliques telles que la mucopolysaccharidose congénitale et la cystinurie : l’évaluation des symptômes systémiques est importante pour le diagnostic différentiel.
Les maladies incluses dans l’ASD (anomalie d’Axenfeld-Rieger, anomalie de Peters, aniridie, dystrophie polymorphe postérieure, microphtalmie, microcornée, etc.) doivent être envisagées mutuellement dans le diagnostic différentiel.3)
QL'anneau embryotoxique postérieur (posterior embryotoxon) isolé peut-il être associé à un glaucome ?
A
Un anneau embryonnaire postérieur isolé (sans symptômes systémiques) se distingue du SRA, mais il peut également être l’une des manifestations associées du SRA. L’anneau embryonnaire postérieur peut être observé dans des yeux sains et ne signifie pas nécessairement un risque de glaucome en soi. Cependant, lorsqu’il est associé à d’autres maladies comme le syndrome d’Alagille, une surveillance de la pression intraoculaire est nécessaire.
Le traitement médicamenteux est un traitement adjuvant visant à abaisser la pression intraoculaire à court terme avant la chirurgie et à contrôler la pression postopératoire. Le choix des médicaments est fondamentalement le même que pour le glaucome à angle ouvert de l’adulte. Cependant, les bêtabloquants nécessitent une attention particulière en cas d’asthme bronchique ou de bradycardie, et des cas d’apnée chez les nouveau-nés ont été rapportés. L’acétazolamide par voie orale (5 à 10 mg/kg toutes les 6 à 8 heures) est également possible.
Le glaucome congénital précoce nécessite fondamentalement un traitement chirurgical. Le traitement médicamenteux est un complément.
Goniotomie : convient comme première intervention pour les cas avec peu d’opacité cornéenne. Elle présente l’avantage de ne pas traumatiser la conjonctive. À l’aide d’un objectif de Barkan ou de Swan-Jacob, on gratte la surface du trabéculum avec un couteau à goniotomie.
Trabéculotomie : réalisable indépendamment de la présence d’opacités cornéennes. Également pratiquée comme intervention complémentaire en cas d’efficacité insuffisante de la goniotomie.
Trabéculectomie et chirurgie de tube de drainage : options en cas d’échec de la chirurgie de l’angle. Dans l’ARS, on choisit la chirurgie de l’angle si l’angle est ouvert et que la couverture du trabéculum par les synéchies antérieures périphériques n’est pas étendue, mais le taux de succès est inférieur à celui du PCG. En cas d’échec de la chirurgie de l’angle, la trabéculectomie ou la chirurgie de tube de drainage avec plaque peuvent être le premier choix. 4)
Dans l’anomalie de Peters, le traitement suit celui du glaucome congénital primitif, mais seulement environ un tiers des cas opérés obtiennent une pression intraoculaire postopératoire satisfaisante, et le pronostic est souvent défavorable. En raison des anomalies cornéennes associées, il est souvent difficile d’obtenir une vision utile. 4)
Le glaucome associé à l’aniridie est traité de la même manière que le PCG. 4)
Anomalie de Peters : Dans les cas légers, l’opacité cornéenne diminue souvent progressivement. Si la pression intraoculaire est normale, une certaine amélioration est fréquente, et comme le pronostic après une greffe de cornée est mauvais, la transplantation cornéenne n’est généralement pas réalisée chez le jeune enfant. De nombreux cas résistent au traitement médicamenteux du glaucome et, même après une chirurgie de reconstruction de la voie d’écoulement, le contrôle est difficile, avec un mauvais pronostic.
CHED : En cas d’insuffisance endothéliale cornéenne, une greffe de cornée (y compris une greffe endothéliale) peut être indiquée.
Cornée sclérosée : peut être associée à d’autres syndromes ASD ; les cas graves peuvent nécessiter une greffe de cornée.
Photophobie et fatigue oculaire : envisager le port de lunettes filtrantes et la prescription de lentilles de contact cosmétiques.
Kératopathie associée à l’aniridie (AAK) : en cas d’opacité cornéenne progressive, une greffe de cellules souches du limbe cornéen peut être envisagée.
Dépistage de la tumeur de Wilms : en cas d’aniridie sporadique, il est recommandé d’orienter le patient vers un pédiatre et d’effectuer un dépistage jusqu’à l’âge de 6 ans. 4)
Même si la pression intraoculaire diminue, un traitement de l’amblyopie est souvent nécessaire. L’anisométropie réfractive, l’astigmatisme irrégulier, l’opacité cornéenne et les stries de Haab peuvent provoquer une amblyopie, il est donc important de poursuivre les examens de l’acuité visuelle et de la réfraction en parallèle de la mesure de la pression intraoculaire. La progression de la myopie et l’allongement de la longueur axiale suggèrent une progression du glaucome, nécessitant des mesures régulières.
6. Physiopathologie et mécanisme détaillé de la maladie
La formation normale du segment antérieur suit un programme de développement complexe. Au début de la troisième semaine de gestation, le sillon optique se forme dans la plaque neurale, marquant le début du développement de l’appareil visuel. À la fin de la troisième semaine, la vésicule optique se forme, et à la quatrième semaine, la cupule optique apparaît. La fermeture de la fissure embryonnaire commence vers la sixième semaine et se termine à la septième semaine. Le mésenchyme recouvrant la face antérieure du cristallin se sépare pour former la chambre antérieure.
Les cellules de la crête neurale se dé-épithélialisent à partir de la crête neurale et migrent vers diverses régions de l’œil après une transition épithélio-mésenchymateuse. Les cellules trabéculaires sont dérivées de la crête neurale, tandis que le tissu conjonctif périvasculaire du canal de Schlemm est dérivé des cellules endothéliales vasculaires, et cette différence d’origine constitue la principale zone de résistance à l’écoulement de l’humeur aqueuse.
PAX6 : « facteur maître » du développement oculaire. Situé sur le chromosome 11. Impliqué dans l’aniridie, l’anomalie de Peters et le syndrome d’ectropion irien congénital.
PITX2 : facteur de transcription. Chromosome 4 (4q25). Associé à la fois aux symptômes oculaires et auditifs observés dans le syndrome d’Axenfeld-Rieger.
FOXC1 : facteur de transcription. Chromosome 6 (6p25). Impliqué dans l’ARS, associé à la fois aux symptômes oculaires et auditifs, comme PITX2.
Le glaucome secondaire dans l’ASDA survient principalement par un mécanisme de dysgénésie de la voie d’écoulement de l’humeur aqueuse. Plus précisément, les facteurs suivants sont impliqués de manière complexe.
Immaturité du trabéculum : le tissu conjonctif juxtacanaliculaire est anormalement épais et la matrice extracellulaire s’accumule de manière excessive.
Adhésion du corps ciliaire au trabéculum : la contraction du muscle ciliaire tire l’éperon scléral vers l’avant, comprimant le canal de Schlemm et le trabéculum.
Insertion haute de la racine de l’iris : la racine de l’iris se trouve au niveau du trabéculum, obstruant l’écoulement de l’humeur aqueuse.
Hypoplasie ou absence du canal de Schlemm.
Le syndrome ICE a une étiologie différente des autres ASDA. Une théorie virale impliquant le virus de l’herpès simplex (HSV) dans la dégénérescence des cellules endothéliales cornéennes a été proposée, mais n’est pas confirmée. Il est acquis, survient chez les adultes d’âge moyen (légèrement plus fréquent chez les femmes) et est généralement unilatéral, ce qui le distingue également des autres ASDA.
Pathologie de la kératopathie associée à l’aniridie (AAK)
Les patients atteints d’aniridie développent une opacité cornéenne progressive tout au long de leur vie. L’insuffisance en cellules souches limbiques (limbal stem cell deficiency, LSCD) est considérée comme le mécanisme principal. Plusieurs études ayant confirmé une mutation PAX6 ont documenté cette évolution progressive. L’incidence rapportée varie de 20 à plus de 80 %, et elle se manifeste souvent de manière symétrique, mais pas toujours. 2)
Les analyses d’exome et de génome entier progressent dans l’identification de nouveaux gènes associés. Le gène causal n’est toujours pas identifié dans 40 à 75 % des cas, et l’analyse des cas non résolus restants constitue un défi important pour l’avenir. L’élucidation de la corrélation entre le génotype et le phénotype clinique est attendue pour des applications en médecine personnalisée.
Chez les patients porteurs de mutations FOXC1 et PITX2, l’âge d’apparition du glaucome et le tableau clinique sont variables. Bien que le génotype puisse être associé à une diversité phénotypique, une même mutation génétique peut se manifester par des formes cliniques différentes, ce qui rend le diagnostic et le pronostic difficiles. 1)
AAK et transplantation de cellules souches limbiques
L’incidence de la kératopathie associée à l’aniridie (AAK) est rapportée entre 20 et plus de 80 %, et plusieurs études avec mutation PAX6 confirmée ont documenté une progression de l’opacité cornéenne tout au long de la vie. La recherche sur la greffe de cellules souches limbiques ciblant le LCSD progresse, mais elle en est encore au stade de la recherche et n’a pas encore été établie comme traitement standard. 2)
Des études épidémiologiques ont montré une association entre l’exposition à la pollution atmosphérique (PM2,5) avant la conception et pendant la grossesse et le risque d’ASDA, et des applications en santé publique sont explorées d’un point de vue de médecine préventive environnementale. Cela pourrait conduire à des stratégies de prévention futures.
Application des dispositifs chirurgicaux mini-invasifs
L’application des lasers à micropulsions et des dispositifs de chirurgie du glaucome mini-invasive (MIGS) chez les enfants atteints d’ASDA est en phase de recherche. Les données à long terme sont limitées, et l’efficacité et la sécurité équivalentes à celles du glaucome adulte ne sont pas établies.
Knight LSW, Ruddle JB, Taranath DA, et al. Childhood and Early Onset Glaucoma Classification and Genetic Profile in a Large Australasian Disease Registry. Ophthalmology. 2021;128(11):1549-1560. doi:10.1016/j.ophtha.2021.04.016.
Hu JCW, Trief D. A narrative review of limbal stem cell deficiency & severe ocular surface disease. Ann Eye Sci. 2023;8:13. doi:10.21037/aes-22-35. https://aes.amegroups.org/article/view/7385/html
Pazos M, Traverso CE, Viswanathan A; European Glaucoma Society. European Glaucoma Society - Terminology and guidelines for glaucoma, 6th Edition. Br J Ophthalmol. 2025;109(Suppl 1):1-212. doi:10.1136/bjophthalmol-2025-egsguidelines. PMID:41026937.