Das keratolimbale Allotransplantat (KLAL) ist eine Art der okulären Oberflächenstammzelltransplantation (OSST) zur Behandlung des Limbusstammzellmangels (LSCD). Dabei wird allogenes Limbusgewebe, das an einem korneoskleralen Träger eines verstorbenen Spenders haftet, transplantiert, um die Homöostase des Hornhautepithels wiederherzustellen.
Bei LSCD sind die epithelialen Stammzellen der Hornhaut geschädigt, und das Hornhautepithel wird durch Bindehautepithel ersetzt 1). Diese Konjunktivalisierung führt zu einem Verlust der Hornhauttransparenz, Sehverschlechterung, Narbenbildung und Neovaskularisation1). Eine alleinige Hornhauttransplantation ist bei LSCD unwirksam, und wiederholte Oberflächendefekte sowie epitheliale Heilungsstörungen führen zum Transplantatversagen 2).
KLAL ist indiziert bei: bilateralem LSCD ohne Lebendspender; unilateralem LSCD, wenn das gesunde Auge als Spender ungeeignet ist; LSCD mit geringer Bindehautbeteiligung wie Aniridie oder kontaktlinsenassoziiertem LSCD.
Historisch berichtete Thoft 1984 über die Hornhautepitheltransplantation. 1990 berichteten Turgeon und Thoft über eine modifizierte Technik mit Limbusgewebe, was das erste dokumentierte KLAL darstellt.
QWas ist der Unterschied zwischen KLAL und Autotransplantat?
A
Das Autotransplantat (CLAu, SLET) entnimmt Limbusgewebe vom gesunden Auge bei unilateralem LSCD, sodass keine Immunsuppression erforderlich ist. Im Gegensatz dazu verwendet KLAL Allotransplantatgewebe eines verstorbenen Spenders, was eine systemische Immunsuppression erfordert, aber auch bei bilateralem LSCD oder wenn das gesunde Auge als Spender ungeeignet ist, durchgeführt werden kann. Eine systematische Übersichtsarbeit ergab eine anatomische Erfolgsrate von 81 % für autologe CLAu und 78 % für SLET2).
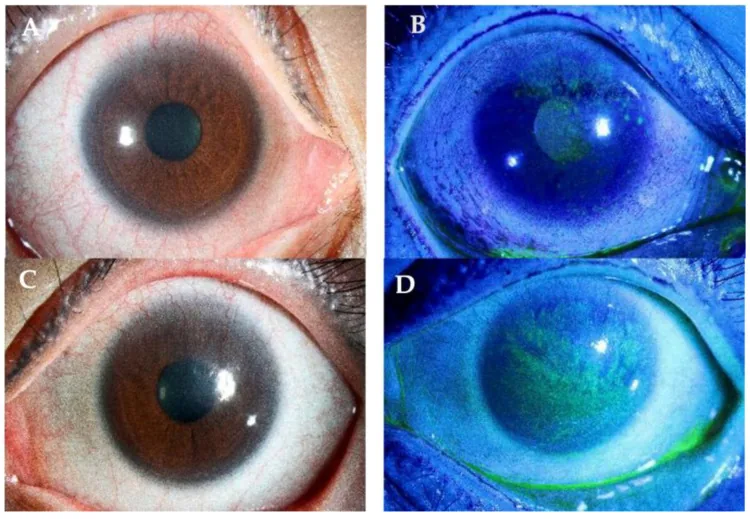
Yhu Fhei Lee, Dayna Wei Wei Yong, Ray Manotosh A Review of Contact Lens-Induced Limbal Stem Cell Deficiency 2023 Dec 5 Biology (Basel). 2023 Dec 5; 12(12):1490 Figure 3. PMCID: PMC10740976. License: CC BY.
A und C zeigen unter Fluoresceinfärbung eine Neovaskularisation, die sich vom Limbus zur Hornhaut erstreckt. B und D zeigen einen Pannus und eine oberflächliche Trübung, die bis zur Sehachse reichen, was das Ausmaß der Limbusstammzellschädigung verdeutlicht.
Patienten mit LSCD klagen hauptsächlich über eine verminderte Sehschärfe. Ursache ist der Verlust der Transparenz der Hornhaut durch Konjunktivalisierung und Narbenbildung. Bei anhaltendem Hornhautepitheldefekt treten Augenschmerzen auf. Auch Photophobie und Tränenfluss werden beobachtet.
Bei leichtem LSCD zeigt die Fluorescein-Färbung eine wirbelartige (whorl-like) Keratopathie1). Bei mäßigem Schweregrad treten oberflächliche Hornhautneovaskularisation und peripherer Pannus auf1). Bei schwerem LSCD ersetzt eine zirkumferenzielle Konjunktivalisierung die gesamte Hornhaut durch Konjunktivalepithel1).
Die Schweregradklassifikation des LSCD basiert auf dem klinischen Grading und unterscheidet leicht (2-4 Punkte), mäßig (5-7 Punkte) und schwer (8-10 Punkte)1). Die basale Epithelzeldichte beträgt in Kontrollaugen 9.252 Zellen/mm², während sie in schweren LSCD-Augen auf 2.821 Zellen/mm² abfällt1).
LSCD entsteht, wenn die limbalen Stammzellen geschädigt werden. Die häufigste Ursache ist die Aniridie (30,9 %), gefolgt von chemischen Verätzungen (20,6 %), Kontaktlinsen (16,8 %) und SJS (10,4 %) 1). Bei einseitigen Fällen sind chemische Verätzungen am häufigsten 1).
Glaukomchirurgie ist ein Risikofaktor für LSCD1). Direkte mechanische Traumata durch Trabekulektomie oder Kammerwasser-Shunt-Operationen schädigen die limbalen Stammzellen 1). Die gleichzeitige Anwendung von Antimetaboliten (Mitomycin C, 5-FU) beeinflusst ebenfalls die limbale Mikroumgebung 1). Es wurde berichtet, dass 35 % der LSCD-Patienten ein Glaukom oder eine okuläre Hypertension aufwiesen 1).
Bei chemischen Verätzungen werden die im Limbus corneoscleralis vorhandenen epithelialen Stammzellen der Hornhaut geschädigt. Das Verschwinden der Vogt-Palisaden spiegelt das Ausmaß der Schädigung der epithelialen Stammzellen der Hornhaut wider.
Die Diagnose einer LSCD basiert auf einer Kombination aus klinischen Befunden und bildgebenden Untersuchungen 1).
Klinische Untersuchungen
Spaltlampenmikroskopie : Ermöglicht die direkte Beobachtung des Verlusts der Vogt-Palisaden 1). Mit Fluorescein-Färbung wird das Ausmaß der spiraligen Epitheliopathie und der Konjunktivalisierung beurteilt 1).
Impressionzytologie : Entnahme des Augenoberflächenepithels mit einem Celluloseacetatfilter 1). Das Vorhandensein von Becherzellen weist auf eine Konjunktivalisierung hin 1). Es gilt als eines der diagnostischen Kriterien für LSCD, aber die Probenqualität ist variabel 1).
Bildgebende Untersuchungen
In-vivo-Konfokalmikroskopie : Ermöglicht die Beobachtung morphologischer Unterschiede zwischen Hornhaut- und Bindehautepithel 1). Das Hornhautepithel besteht aus großen, polygonalen, abgeflachten Zellen, während das Bindehautepithel kubisch mit einem stark reflektierenden Zytoplasma ist 1).
Vorderabschnitts-OCT (AS-OCT) : Ermöglicht eine schnelle, nicht-invasive Beurteilung 1). Das Ausmaß der Konjunktivalisierung der Hornhaut und Veränderungen der Epitheldicke können beurteilt werden.
Die Operation wird unter Vollnarkose oder retrobulbärer Anästhesie durchgeführt. Es erfolgt eine 360°-limbale Konjunktivalinzision (Peritomie), und das fibrovaskuläre Pannus sowie das abnorme Epithel werden von der Hornhautoberfläche entfernt. Aus zwei Spender-Korneoskleralringen wird die zentrale Hornhaut exzidiert, um vier sichelförmige Limbusgewebestücke (Kreszenten) herzustellen. Die hintere Hälfte jedes Kreszenten wird durch lamelläre Dissektion entfernt, und in der Regel werden drei Kreszenten um die Hornhaut des Empfängers platziert. Sie werden mit 10-0-Nylonfäden und Gewebekleber fixiert.
Die Auswahlkriterien für Spendergewebe empfehlen junge Spender unter 60 Jahren, eine Minimierung der Zeit zwischen Tod und Konservierung sowie eine Transplantation innerhalb von 5 Tagen nach dem Tod.
Für das langfristige Überleben des Transplantats nach KLAL ist eine systemische Immunsuppression unerlässlich. Nach dem Cincinnati-OSSTP-Protokoll werden orales Tacrolimus (4 mg zweimal täglich) und MMF (1 g zweimal täglich) 1–2 Wochen vor der Operation begonnen. Postoperativ wird orales Prednison (1 mg/kg/Tag) hinzugefügt und über 1–3 Monate ausgeschlichen. Bei Patienten mit stabiler Augenoberfläche wird Tacrolimus ab 12–18 Monaten postoperativ und MMF ab 3 Jahren schrittweise reduziert.
Stabile Augenoberfläche : Wird bei 73–77 % der Patienten unter geeigneter Dreifach-Immunsuppression erreicht. Dies wird bei einer durchschnittlichen Nachbeobachtungszeit von 4,5–4,7 Jahren berichtet.
Klassifikation des Transplantatversagens: Primärversagen (fehlende Epithelisierung innerhalb der ersten postoperativen Woche), Teilversagen (Mischung aus Konjunktivalisierung und gesundem Epithel), Totalversagen (zirkumferenzielles Rezidiv der LSCD), Spätversagen (durch chronische Abstoßung).
CLET (kultivierte Limbus-Epithel-Transplantation): Anatomische Erfolgsrate 61,4 %, funktionelle Erfolgsrate 53 %, unterlegen gegenüber CLAu und SLET2).
QWie lange ist eine Immunsuppression erforderlich?
A
Die systemische Immunsuppression wird in der Regel schrittweise reduziert: Tacrolimus ab 12–18 Monaten postoperativ, MMF ab 3 Jahren. Wenn die Augenoberfläche stabil ist und keine Abstoßung in der Vorgeschichte vorliegt, kann sie schließlich abgesetzt werden. Bei Abstoßungsanamnese muss jedoch eine niedrig dosierte Immunsuppression unbegrenzt fortgesetzt werden. Eine Zusammenarbeit mit einem Organtransplantationsspezialisten wird empfohlen.
QVerbessert sich das Sehvermögen nach KLAL?
A
Das Hauptziel der KLAL ist die Etablierung einer stabilen Augenoberfläche, nicht die direkte Sehverbesserung. Nach Stabilisierung der Hornhautoberfläche kann jedoch durch eine optische Hornhauttransplantation (PKP oder DALK) eine Sehverbesserung erwartet werden. In einer Fallserie von DMAK + allo-SLET nach KLAL verbesserte sich der korrigierte Visus von 20/200 auf 20/403).
Die Limbus-Stammzellen befinden sich in den Palisaden von Vogt in der Basalschicht des Limbus1). Diese Stammzellen wandern zentripetal, differenzieren sich zunächst zu basalen Epithelzellen, proliferieren dann und wandern zur Oberfläche, wo sie abgeschilfert werden1). Der Limbus fungiert auch als Barriere gegen das Eindringen der Konjunktiva in die Hornhaut1).
Bei LSCD führt die Schädigung der Limbus-Stammzellen dazu, dass das Hornhautepithel durch Konjunktivalepithel ersetzt wird und die Hornhauttransparenz verloren geht. Wenn nur 7 % der Limbus-Stammzellen erhalten sind, kann mit modernen chirurgischen Techniken das Hornhautepithel regeneriert werden. 1)
Bei chronischen Augenerkrankungen verändert die kombinierte Wirkung von Entzündungszytokinen und Schleimhautpemphigoid die Mikroumgebung der limbalen Stammzellen 1). Bei LSCD in Verbindung mit bullöser Keratopathie dauert die Epithelisierung etwa 6 Tage in Augen ohne Hornhautneovaskularisation, verglichen mit etwa 29 Tagen in Augen mit Neovaskularisation1).
Das Hornhautepithel und das Bindehautepithel unterscheiden sich morphologisch 1). Das normale Hornhautepithel besteht aus großen, polygonalen Plattenepithelzellen, wobei die Basalschicht ein hyporeflektives Zytoplasma und klare Grenzen aufweist 1). Das Bindehautepithel ist kubisch mit einem hyperreflektiveren Zytoplasma und enthält Becherzellen 1). Zytokeratin 3 ist spezifisch für das Hornhautepithel, während Zytokeratin 19 als spezifisch für das Bindehautepithel gilt 1).
Als Rettungsmaßnahme nach KLAL-Versagen wurde die Kombination aus anteriorer lamellärer Keratoplastik mit azellulärer Descemet-Membran (DMAK) und allogener vereinfachter limbaler Epitheltransplantation (allo-SLET) berichtet 3). Die Descemet-Membran ist resistenter gegen Abbau als die humane Amnionmembran (HAM): Sie hält über 24 Stunden in hochkonzentrierter Kollagenase, während HAM innerhalb von 30 Minuten abgebaut wird 3).
Auf Descemet-Membran kultivierte limbale Stammzellen zeigten eine bessere Expression von Stammzellmarkern (ABCG2, p63) im Vergleich zu HAM 3). In einem Fall von LSCD im Zusammenhang mit autoimmunem polyglandulärem Syndrom, nach KLAL-Abstoßung und Behandlung mit DMAK + allo-SLET, verbesserte sich der bestkorrigierte Visus von 20/200 auf 20/40, und es trat 1,5 Jahre lang kein Rezidiv des Epitheldefekts auf 3).
Die Descemet-Membran bietet Vorteile hinsichtlich Transparenz, Haltbarkeit und als Substrat für die limbale Stammzellkultur und ist eine vielversprechende Rettungsoption nach KLAL-Versagen. Langzeitwirksamkeit und Immunantwort bedürfen jedoch weiterer Untersuchungen. 3)
Auch bei der Diagnose von LSCD wurden Fortschritte erzielt 1). Die verbesserte Genauigkeit der In-vivo-Konfokalmikroskopie und der AS-OCT ermöglicht eine objektive Beurteilung des Schweregrads der LSCD und eine bessere Überwachung des Behandlungserfolgs 1). Bei Aniridie könnten Fortschritte in der genetischen Analyse zur Prognosevorhersage und zum Management beitragen 1).