Das Stevens-Johnson-Syndrom (SJS) ist eine akute Erkrankung der Haut und Schleimhäute, die nach plötzlichem hohem Fieber, Konjunktivitis und Hautausschlag zu Erosionen und Blasenbildung an der gesamten Haut und den Schleimhäuten führt. Die toxische epidermale Nekrolyse (TEN) ist eine Krankheitsform, die schwere Formen des SJS umfasst. Viele Fälle haben eine Vorgeschichte von Medikamenteneinnahme vor Ausbruch der Erkrankung; es handelt sich auch um eine schwerwiegende Arzneimittelnebenwirkung. Die Erkrankung wurde nach den US-amerikanischen Kinderärzten Albert Mason Stevens und Frank Chambliss Johnson benannt, die 1922 zwei Kinder mit Fieber, Konjunktivitis sowie Erosionen der Haut und Mundschleimhaut beschrieben. Später, im Jahr 1956, berichtete Alan Lyell über die toxische epidermale Nekrolyse (TEN), und beide wurden als dasselbe Spektrum schwerer Arzneimittelexantheme eingeordnet.
Klassifikation
Fläche der Hautläsionen
SJS
weniger als 10 % der Körperoberfläche
SJS/TEN-Überlappung
10–30 % der Körperoberfläche
TEN
mehr als 30 % der Körperoberfläche
Die Augenmanifestationen von SJS und TEN sind ähnlich, und eine Unterscheidung allein anhand der Augenmerkmale ist schwierig. Daher werden SJS und TEN in der Augenheilkunde häufig zusammenfassend als SJS im weiteren Sinne bezeichnet. Auch der Pathomechanismus und die Augenspätfolgen werden als weitgehend gemeinsames Spektrum betrachtet.
Die Inzidenz beträgt wenige Fälle pro 1 Million Einwohner pro Jahr, was die Erkrankung sehr selten macht, sie tritt jedoch in jedem Alter ohne Geschlechtsunterschied auf, einschließlich bei Kindern. In den USA wird eine Inzidenz von 12,35 Fällen pro 1 Million Einwohner pro Jahr berichtet 8). Die Letalität ist mit 4,8 % für SJS und 14,8 % für TEN hoch 3), und für TEN wird sogar eine Letalität von bis zu 30 % berichtet 1).
Die Häufigkeit von Augenkomplikationen bei SJS/TEN liegt bei etwa 70 %, bei Erwachsenen treten sie in 53–88 % der Fälle auf 8). Insbesondere schwere Augenkomplikationen mit sowohl Pseudomembranen als auch Hornhaut- und Bindehautepitheldefekten treten bei etwa 40 % aller SJS/TEN-Fälle auf 10). Aufgrund der hohen Letalität steht bei Ausbruch der Erkrankung die systemische Behandlung im Vordergrund, aber die häufigste Spätfolge ist eine Augenschädigung, die zu Sehbehinderung durch schwere Hornhauttrübung und trockenem Auge führt, die lebenslang anhalten 11). Bei Fällen, die zu einer Erschöpfung der Hornhautepithelstammzellen führen, fehlen die normalerweise vom Limbus zentripetal gelieferten Hornhautepithelzellen, und bindehautstämmiges Gewebe mit Gefäßen dringt in die Hornhautoberfläche ein. Da diese schwer reversible Veränderung den Kern der Sehbehinderung ausmacht, beeinflusst die Qualität der Akutbehandlung innerhalb weniger Tage nach Ausbruch die Prognose maßgeblich 13).
SJS/TEN wird häufig in der Pädiatrie, Dermatologie und Notfallmedizin erkannt, aber in Fällen, in denen die Augensymptome den Hautveränderungen vorausgehen, kann der Patient auch erstmals einen Augenarzt aufsuchen. Bei einem Krankheitsbild mit generalisiertem Fieber und Hautausschlag ist die Abgrenzung zu Erkrankungen mit akuter beidseitiger Konjunktivitis stets problematisch. Daher ist es auch für den allgemeinen Augenarzt wichtig, eine frühzeitige Diagnose und die Überleitung zu einer multidisziplinären Behandlung im Rahmen einer interprofessionellen Zusammenarbeit im Blick zu haben. Bei der Erstuntersuchung sollten die Beurteilung der Allgemeinsymptome und die Inspektion der Haut und Schleimhäute nicht vernachlässigt werden; bei beidseitiger Konjunktivitis mit Fieber ist eine aktive Zusammenarbeit mit der Dermatologie und Notfallmedizin erforderlich. Bei Verdachtsfällen sollte frühzeitig eine stationäre Behandlung eingeleitet werden, und in der Augenheilkunde verbessert eine zügige und aktive Intervention in der Akutphase die Sehprognose direkt.
QWas ist der Unterschied zwischen SJS und TEN?
A
SJS und TEN sind Erkrankungen desselben Spektrums und werden nach der Fläche der Hautveränderungen klassifiziert. Weniger als 10 % der Körperoberfläche entsprechen SJS, 10–30 % SJS/TEN-Überlappung und mehr als 30 % TEN. Die Augenmanifestationen sind bei beiden ähnlich und schwer zu unterscheiden, daher werden sie in der Augenheilkunde zusammenfassend als SJS im weiteren Sinne bezeichnet. Die Letalität beträgt 4,8 % für SJS und 14,8 % für TEN 3). Die Inzidenz schwerer Augenkomplikationen liegt bei etwa 40 % aller SJS/TEN-Fälle 10).
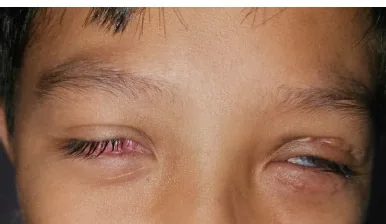
Wibowo E, Maharani RV, Sutikno NA. Symblepharon as Ocular Manifestation Post Stevens-Johnson Syndrome: A Rare Case. Romanian Journal of Ophthalmology. 2024 Oct-Dec; 68(466):$2. Figure 1. PMCID: PMC11809831. License: CC BY.
Beide Augen sind verengt und können nicht vollständig geöffnet werden; das rechte vordere Augenfeld ist von Narben und Granulationsgewebe bedeckt, das linke Auge zeigt Verengungen und Narben nach Lidtransplantation. Dies zeigt schwere chronische Veränderungen der Augenoberfläche bei Hauptsymptomen und klinischen Befunden.
Rötung, Fremdkörpergefühl und Augenschmerzen beider Augen: Gleichzeitig mit den Augensymptomen oder innerhalb weniger Tage treten hauptsächlich am Rumpf Hautausschläge auf.
Sehverschlechterung: Verursacht durch Hornhaut- und Bindehautepitheldefekte in der akuten Phase und Hornhauttrübungen in der chronischen Phase.
Trockenheitsgefühl und Lichtempfindlichkeit: Halten in der chronischen Phase aufgrund von trockenem Auge an.
Müdigkeit und Halsschmerzen: Werden von vielen Patienten als grippeähnliche Prodromalsymptome vor Ausbruch der Erkrankung wahrgenommen.
Klinische Befunde (vom Arzt bei der Untersuchung festgestellte Befunde)
Beidseitige Bindehautrötung: Schwere Rötung, die fast gleichzeitig mit Schleimhaut- und Hautausschlägen auftritt.
Pseudomembranbildung: Charakteristischer Befund der akuten Phase, der flügelartig an der Bindehautoberfläche haftet.
Hornhaut- und Bindehautepitheldefekte: Können großflächig sein. Anhaltende Epitheldefekte können zu Hornhautinfektionen, Hornhauteinschmelzung oder -perforation führen.
Rötung und Schwellung der Augenlider: In schweren Fällen kann das Auge nicht einmal geöffnet werden. Auch Wimpernverlust wird beobachtet.
Hornhautpseudomembran: Kann einige Tage nach Krankenhausaufenthalt auftreten4).
Augenbefunde der akuten Phase stehen in direktem Zusammenhang mit der Schweregradeinteilung: Die akute Augenschweregradeinteilung nach Sotozono et al. unterteilt in Grade 0 bis 3 basierend auf dem Vorhandensein von Pseudomembran, Epitheldefekt und Hornhautepitheldefekt11).
Wenn Patienten vor dem Auftreten von Hautausschlägen einen Augenarzt aufsuchen, kann dies fälschlicherweise als virale Bindehautentzündung diagnostiziert werden. Das Vorhandensein von Fieber und generalisierten Hautausschlägen muss immer überprüft werden.
Chronische Phase (Narbenstadium)
Schweres trockenes Auge: Fast alle Fälle weisen eine Tränensekretionsinsuffizienz aufgrund einer Verstopfung der Tränendrüsenausführungsgänge auf. Häufig liegt auch eine Meibom-Drüsen-Dysfunktion vor.
Trichiasis: Besteht über Jahre und verschlechtert den Zustand der Augenoberfläche. Oft ist eine Entfernung der Wimpern 3- bis 4-mal pro Monat im Zyklus erforderlich.
Symblepharon: Der Fornix verschwindet und die Lidbindehaut verwächst mit der Bulbusbindehaut.
Konjunktivalisierung der Hornhaut: Wenn die limbalen Stammzellen des Hornhautepithels verloren gehen, bedeckt das Bindehautgewebe die Hornhautoberfläche und führt zu Sehstörungen.
Epitheliale Keratinisierung: In schweren Fällen verhornt die Oberfläche von Hornhaut und Bindehaut wie Haut.
Verlust der Palisaden von Vogt: Dies ist ein klinisches Zeichen für den Verlust der limbalen Stammzellen des Hornhautepithels.
Die Schwere der Augenkomplikationen korreliert nicht unbedingt mit dem Ausmaß der Hautläsionen. Es wurden Fälle berichtet, bei denen ein Auge, das nicht mit Augentropfen behandelt wurde, schwerere Schäden erlitt als das kontralaterale Auge, was darauf hindeutet, dass SJS/TEN grundsätzlich eine systemische Immunerkrankung ist1).
Für die Vorhersage von Augenkomplikationen sind das Vorhandensein von Pseudomembranen, Hornhautepitheldefekten und Bindehautrötung in der akuten Phase wichtige Faktoren. Eine japanische multizentrische Studie berichtete, dass bei Patienten mit Pseudomembranbildung oder Hornhautepitheldefekten in der akuten Phase schwere chronische Augenfolgen häufiger auftreten11). Daher müssen die Augenuntersuchungen in der akuten Phase täglich oder alle paar Tage detailliert dokumentiert und die Graduierung im Zeitverlauf verfolgt werden.
SJS/TEN wird häufig durch die Verabreichung von Medikamenten ausgelöst. Bei Kindern geht häufig eine Mykoplasmeninfektion voraus. Vor Ausbruch der Erkrankung treten oft grippeähnliche Symptome wie Müdigkeit und Halsschmerzen auf, und es wird angenommen, dass eine Virusinfektion als Auslöser dient, aber der genaue Mechanismus ist unbekannt. Die Latenzzeit zwischen Beginn der Medikamenteneinnahme und Ausbruch beträgt meist relativ kurz, etwa 4 Tage bis 1 Monat, wobei der häufigste Zeitraum 2 bis 3 Wochen nach Beginn der Einnahme des verdächtigen Medikaments ist. Es gibt jedoch auch Fälle nach längerer Einnahme, sodass ein kausaler Zusammenhang allein aufgrund der Einnahmedauer nicht ausgeschlossen werden kann.
Ursachenkategorie
Typische Medikamente/Faktoren
Antibiotika
Sulfonamide (z. B. Cotrimoxazol) am häufigsten
Antiepileptika
Carbamazepin, Phenytoin, Lamotrigin
Analgetika und Antipyretika
Nichtsteroidale Antirheumatika (NSAR)
Medikamente gegen Hyperurikämie
Allopurinol
Antitumormittel
Immun-Checkpoint-Inhibitoren (ICI)
Infektionen
Mykoplasmen-Infektion (besonders bei Kindern), Herpes-simplex-Virus
Insbesondere wurden zahlreiche Fälle durch Antiepileptika berichtet. Es wurden Fälle beschrieben, die 35 Tage nach der Kombination von Carbamazepin und Phenytoin auftraten 3), sowie Fälle, bei denen nach Umstellung auf Lamotrigin ein TEN auftrat 7). Der Beitrag von NSAR zur Entstehung wird durch die Hemmung der Prostaglandinsynthese als Mechanismus vermutet.
In den letzten Jahren wurden auch SJS/TEN durch Immun-Checkpoint-Inhibitoren (ICI) berichtet. In einer Post-Marketing-Studie zu Tislelizumab wurden unter 3.795 unerwünschten Ereignissen 3 Fälle von TEN identifiziert 5). Die mediane Zeit bis zum Auftreten von ICI-induziertem SJS/TEN beträgt 32 Tage, und in einer Nachbeobachtung von 305 Fällen starben 69 Patienten 4).
Auch Augentropfen können SJS/TEN auslösen. Es wurde ein Fall berichtet, bei dem Brinzolamid-Augentropfen, ein Sulfonamid-Carboanhydrasehemmer, ein systemisches SJS/TEN-Overlap (Körperoberfläche 99%) verursachten 1). Es wird angenommen, dass die systemische Absorption über die Bindehaut und Nasenschleimhaut bei genetisch prädisponierten Personen eine systemische Immunreaktion auslösen kann.
Auch SJS/TEN nach COVID-19-Impfung wurde berichtet 2). Impfstoff-induziertes SJS/TEN tritt tendenziell innerhalb von 1–8 Tagen auf, kürzer als medikamenteninduziertes (2–3 Wochen) 2).
Der HLA-Genotyp ist stark mit der Anfälligkeit für SJS/TEN assoziiert. Vor der Gabe von Allopurinol wird ein Test auf HLA-B58:01 empfohlen, vor Carbamazepin ein Test auf HLA-B15:02 8). Bei japanischen SJS/TEN-Patienten mit schweren Augenkomplikationen wurde zudem eine starke Assoziation mit HLA-A*02:06 berichtet 12). Die Analyse dieser HLA-Polymorphismen spielt eine wichtige Rolle bei der Identifizierung des auslösenden Medikaments und beim Screening vor der Medikamentengabe.
QWelche Medikamente verursachen SJS/TEN?
A
Die häufigsten auslösenden Medikamente sind Sulfonamide (Antibiotika) und Antiepileptika (Carbamazepin, Phenytoin, Lamotrigin), gefolgt von NSAR und Allopurinol. In letzter Zeit gibt es auch zunehmend Berichte über Fälle unter Immun-Checkpoint-Inhibitoren (ICI) 5)6). Auch Augentropfen (Brinzolamid) können systemisches SJS/TEN auslösen 1). Fälle nach COVID-19-Impfung wurden ebenfalls berichtet 2). Bestimmte HLA-Polymorphismen wie HLA-B58:01, HLA-B15:02 und HLA-A*02:06 bestimmen die Anfälligkeit 8)12).
SJS-Diagnosekriterien (3 obligatorische Kriterien)10): (1) Schwere Schleimhautläsionen an den Übergängen von Haut und Schleimhaut (hämorrhagisch oder hyperämisch), (2) Erosionen oder Blasen auf weniger als 10 % der Körperoberfläche, (3) Fieber ≥ 38 °C. Die Diagnose SJS wird gestellt, wenn alle drei Hauptkriterien erfüllt sind. Nebenbefunde umfassen atypische Zielscheiben-Erytheme, beidseitige unspezifische Konjunktivitis (mit oder ohne Hornhautepithelschäden und Pseudomembranbildung) sowie epidermale Nekrose in der Histopathologie.
TEN-Diagnosekriterien (3 obligatorische Kriterien)10): Blasen, Epidermisablösung oder Erosionen auf mehr als 10 % der Körperoberfläche, Ausschluss eines Staphylococcal Scalded Skin Syndrome (SSSS) und Fieber ≥ 38 °C.
Auch bei initial als SJS diagnostizierten Fällen kann ein Übergang in TEN in der Akutphase erfolgen, sodass nach der Erstbeurteilung eine erneute Bewertung erforderlich ist 10).
Graduierung der Augenkomplikationen
Grad 0: Keine Augenveränderungen. Prophylaktische künstliche Tränen 11).
Grad 1: Nur konjunktivale Hyperämie, keine Hornhautläsionen. Antibiotika-Augentropfen 3-mal täglich, Steroid-Augentropfen 6-mal täglich 8).
Grad 2: Hornhautbeteiligung, keine Pseudomembran. Zusätzlich zur oben genannten Behandlung wird eine Amnionmembrantransplantation (Prokera oder AMT) in Betracht gezogen 8).
Grad 3: Hornhautbeteiligung mit Pseudomembran. Eine aggressive Behandlung einschließlich AMT wird in Betracht gezogen 8).
Beurteilung des systemischen Schweregrads mittels SCORTEN: SCORTEN (SCORe of Toxic Epidermal Necrosis) ist ein weit verbreitetes Bewertungssystem zur Prognose von SJS/TEN. Es bewertet sieben Faktoren: Alter ≥40 Jahre, Vorliegen einer malignen Erkrankung, Herzfrequenz ≥120/min, Hautablösungsfläche ≥10%, erhöhter Serumharnstoffstickstoff, erhöhter Blutzucker und erniedrigtes Serumbicarbonat. Mit steigendem Score erhöht sich die Mortalität, und der Score dient als objektiver Indikator für die Notwendigkeit einer Intensivpflege und systemischen Behandlung. Augenärzte tragen zur Therapieentscheidung bei, indem sie mit Dermatologen und Notfallmedizinern zusammenarbeiten und den SCORTEN teilen.
Virale Konjunktivitis: Kann in der akuten Phase bei Patienten, die nur mit Augensymptomen vorstellig werden, fehldiagnostiziert werden. Achten Sie auf systemisches Fieber und Hautausschlag.
Ophthalmisches Narbenpemphigoid (okuläres vernarbendes Pemphigoid): Ähnelt der chronischen Phase der vernarbenden Keratokonjunktivitis. Es handelt sich um eine Autoimmunerkrankung mit schleichendem Verlauf; Impressionzytologie und Immunhistochemie sind zur Differenzierung hilfreich.
Staphylococcal Scalded Skin Syndrome (SSSS): Muss gemäß den TEN-Diagnosekriterien ausgeschlossen werden. SSSS tritt hauptsächlich bei Kindern auf, die Hautablösung erfolgt auf Höhe des Stratum granulosum (oberflächlich) und wird histopathologisch von TEN unterschieden, die eine Nekrose bis zur Dermis verursacht.
Limbusstammzellinsuffizienz nach chemischen oder thermischen Verletzungen: Differenzierung durch Anamnese 8).
Akute hämorrhagische Konjunktivitis: Ähnelt der akuten SJS als virale beidseitige Konjunktivitis, kann aber durch das Fehlen von systemischem Fieber und Hautausschlag unterschieden werden.
Blepharitis/Meibom-Drüsen-Entzündung: Ist als chronische Lidrandveränderung differenzialdiagnostisch zu erwägen, wird aber durch das Vorhandensein oder Fehlen von Narbenbildung unterschieden.
Systemische Behandlung: Eine Steroid-Pulstherapie (Methylprednisolon 500–1.000 mg/Tag für 3 Tage, 1–2 Zyklen) wird früh nach Krankheitsbeginn durchgeführt10). In schweren Fällen werden IVIG-Therapie (0,4–1,0 g/kg/Tag für 3–5 Tage) und Plasmapherese ergänzt. Eine frühzeitige hochdosierte Gabe, sofern der Allgemeinzustand es erlaubt, ist wichtig, um das Fortschreiten der Augenoberflächenentzündung zu hemmen. Optionen wie Cyclosporin (3–5 mg/kg/Tag), Etanercept, Infliximab und andere TNF-α-Inhibitoren können hinzugefügt werden; insbesondere bei steroidresistenten oder persistierenden Fällen wurde eine Wirksamkeit berichtet. Im systemischen Management sind Temperaturkontrolle, Flüssigkeitsmanagement, Ernährungsmanagement und Infektionsprävention auf einer Verbrennungsstation oder Intensivstation unerlässlich, und eine multidisziplinäre Teambehandlung wird durchgeführt.
Lokale Augenbehandlung: Betamethason-Augentropfen oder -Salbe werden 6- bis 10-mal täglich häufig appliziert, um die Bildung von Symblephara zu verhindern. Zur Infektionsprophylaxe werden gleichzeitig antibiotische Augentropfen verwendet. Die Erhaltung der kornealen epithelialen Stammzellen ist der wichtigste Faktor für die Sehprognose in der chronischen Phase11)13).
Frühe Amnionmembrantransplantation (AMT): Wird ab Grad 2 in Betracht gezogen8). Es wurde berichtet, dass bei 9 von 10 extrem schweren Fällen ein BCVA von 20/20 erreicht wurde8). Da auch bei 35 % eine infektiöse Keratitis auftritt, ist eine zusätzliche antibiotische Prophylaxe erforderlich8). Eine augenärztliche Beurteilung innerhalb von 24–48 Stunden nach Beginn und eine frühe AMT-Intervention verbessern die Sehprognose13). Bei der Amnionmembrantransplantation gibt es die Methode der vollständigen Abdeckung der Bulbärkonjunktiva, Fornices und Lidkonjunktiva durch Naht sowie die Verwendung eines nahtlosen ringförmigen Geräts wie Prokera. Prokera kann im Behandlungsraum eingesetzt werden und vermeidet invasive chirurgische Eingriffe in der Akutphase, was seine klinische Anwendung erweitert. Die Amnionmembran hat entzündungshemmende, antiangiogene und anti-narbenbildende Eigenschaften und fördert die Wundheilung der Augenoberfläche durch die Freisetzung von Wachstumsfaktoren und entzündungshemmenden Zytokinen.
Chronische Phase-Behandlung
Trockenes-Auge-Management: Häufige Einträufelung von konservierungsmittelfreien künstlichen Tränen, Hyaluronsäure-Augentropfen, Rebamipid-Augentropfen (Steigerung der Mucinproduktion und entzündungshemmende Wirkung), Diquafosol-Augentropfen (Förderung von Wasser- und Mucinsekretion), zusätzlich Tränenpunkt-Plugs. Das trockene Auge in der chronischen Phase des SJS ist nicht nur eine einfache Verminderung des Tränenvolumens, sondern eine komplexe schwere Form des trockenen Auges mit Lipid-schicht-Anomalien durch Meibom-Drüsen-Dysfunktion, Mucinmangel durch Becherzellverlust und Keratinisierung des Bindehautepithels. Daher ist die Behandlung mit nur einem Medikament schwierig, und eine Mehrfachmedikamentenstrategie zur Ergänzung der einzelnen Tränenfilmkomponenten ist erforderlich. Da Konservierungsmittel die Hornhaut- und Bindehautepithelschädigung verschlimmern, sollten alle Augentropfen möglichst konservierungsmittelfrei gewählt werden.
Management von Trichiasis: Regelmäßige Epilation alle 3–4 Wochen entsprechend dem Haarzyklus. Für feine, depigmentierte Wimpern sind Titanpinzetten oder Nahtpinzetten wirksam. Bei rezidivierenden Fällen werden eine Augenlidoperation zur Wimpernwurzelresektion oder Elektrolyse der Wimpern (Wurzelzerstörung) hinzugefügt. Die Trichiasis nach SJS unterscheidet sich von der gewöhnlichen altersbedingten Entropium; sie wird durch eine leichte marginale Entropium am hinteren Lidrand verursacht, die auf das Narbengewebe der Wimpernwurzel übergreift und die Wuchsrichtung der Wimpern verändert. Daher ist nicht nur eine einfache Epilation, sondern auch eine aktive Behandlung der zugrunde liegenden Lidrandvernarbung erforderlich. Da eine langjährige unbehandelte Trichiasis zu Pterygium pseudocorneale, Hornhautastigmatismus und Hornhauttrübung führen kann, ist eine frühzeitige Intervention wünschenswert.
Entzündungsmanagement: Niedrig dosierte Steroid-Augentropfen zur Unterdrückung der chronischen Entzündung und Verlangsamung des Fortschreitens narbiger Veränderungen. Achtung auf MRSA/MRSE-Kolonisation, Auswahl geeigneter Antibiotika basierend auf der Kultur. Bei SJS/TEN-Patienten mit schweren okulären Spätfolgen ist die Kolonisationsrate von MRSA/MRSE im Bindehautsack hoch, was zu einem Wiederaufflammen der Augenoberflächenentzündung oder infektiöser Keratitis führen kann. Daher werden regelmäßige Bindehautsackkulturen und bei Bedarf der Einsatz von gegen MRSA wirksamen Antibiotika wie Vancomycin oder Linezolid in Betracht gezogen. Bei chronischer Steroid-Augentropfenanwendung ist auf Augeninnendruckerhöhung und sekundäres Glaukom zu achten, regelmäßige Augeninnendruckmessungen durchführen.
Sklerallinsen und Sehrehabilitation: Limbusgestützte harte Kontaktlinsen oder Sklerallinsen sind bei schwerem trockenem Auge und unregelmäßiger Augenoberfläche zur Verbesserung der Sehfunktion wirksam.
Sklerallinsen zur Sehverbesserung: Bei schwerem trockenem Auge mit unregelmäßiger Hornhautoberfläche sind limbusgestützte großformatige Kontaktlinsen wie Sklerallinsen oder BostonSight PROSE (Prosthetic Replacement of the Ocular Surface Ecosystem) nützlich. Durch die Aufrechterhaltung einer Pufferflüssigkeit zwischen Linse und Hornhaut werden feine Hornhautunregelmäßigkeiten optisch korrigiert und ein anhaltendes feuchtes Milieu gewährleistet, wodurch das Fortschreiten der Epithelschädigung gehemmt werden kann. Bei vielen bilateralen SJS-Patienten wird nicht nur eine Refraktionskorrektur, sondern auch eine Linderung von Schmerzen, Photophobie und Fremdkörpergefühl erreicht.
Bei schwerer Sehbehinderung durch Eindringen von Bindehautgewebe in die Hornhaut werden Limbusstammzelltransplantation (limbal stem cell transplantation; LSCT) oder transplantierte Schleimhautepitheltransplantation durchgeführt. Es gibt drei Arten der Limbusstammzelltransplantation je nach Operationsmethode. Ein systematischer Review zur autologen Limbusstammzelltransplantation berichtet eine anatomische/funktionelle Erfolgsrate von 81%/74,4% für das konjunktivale Limbusautotransplantat (conjunctival-limbal autograft; CLAu), 78%/68,6% für die einfache Limbusepitheltransplantation (simple limbal epithelial transplantation; SLET) und 61,4%/53% für die kultivierte Limbusepitheltransplantation (cultivated limbal epithelial transplantation; CLET)9). CLAu und SLET zeigten signifikant bessere Ergebnisse als CLET (p=0,0048)9).
Bei der transplantierten Schleimhautepitheltransplantation gibt es eine Methode unter Verwendung von autologen Mundschleimhautepithelblättern, und die autologe Mundschleimhautepitheltransplantation ist in Japan als fortschrittliche medizinische Behandlung anerkannt13). Bei Fällen wie bilateralem SJS, bei denen kein autologes Limbusgewebe gewonnen werden kann, wird eine kleine Menge der eigenen Mundschleimhaut des Patienten entnommen, in einem Kulturmedium zu einem Epithelblatt expandiert und dann mit Amnionmembran oder Fibrinkleber als Träger auf die Hornhautoberfläche transplantiert. Bei der Herstellung des kultivierten Epithelblatts wird ein mit Retinol und EGF angereichertes Medium verwendet, um xenogene Substanzen auszuschließen. Manchmal wird auch die Hemmung der Fibroblastenaktivität durch Mitomycin C kombiniert. Nach der Transplantation werden Steroid-Augentropfen und Immunsuppressiva fortgesetzt, um ein Wiederaufflammen der Augenoberflächenentzündung zu verhindern.
Eine perforierende Keratoplastik (penetrating keratoplasty; PKP) wird bei Hornhautnarben in Betracht gezogen, aber bei Verlust der Limbusstammzellen kommt es häufig zu Epithelheilungsstörungen, und die Prognose allein ist begrenzt8). Daher wird bei schwerem LSCD zunächst die Augenoberfläche durch Limbusstammzelltransplantation oder transplantierte Schleimhautepitheltransplantation stabilisiert, und anschließend wird häufig eine zweistufige Operation mit lamellärer Keratoplastik oder perforierender Keratoplastik bei Hornhauttrübung durchgeführt13).
Die Boston-Keratoprothese (Boston keratoprosthesis; KPro) wird als Option für schwere Fälle eingesetzt, bei denen andere Behandlungen keine Sehverbesserung ermöglichen 8). Insbesondere bei bilateralem SJS ohne autologe Transplantatquelle wird der Typ II (mucous membrane-covered) mit einem optischen Teil und einem Gewebeteil aus Titan als Mittel zur Überwindung chronischer Konjunktivalisierung und Lidverwachsungen beschrieben. Allerdings sind langfristige Komplikationen wie Glaukom, Netzhautablösung, Infektion und Geräteexposition nicht selten, sodass eine lebenslange strenge Überwachung erforderlich ist. Bei einem Brinzolamid-induzierten SJS/TEN-Fall wurde eine PKP bei Symblepharon und Hornhautnarben durchgeführt, was zu einer Sehschärfe von 0,05 führte 1).
Da Augenoperationen wie Kataraktchirurgie eine Entzündung der Augenoberfläche auslösen können, ist auch bei milden Fällen eine ausreichende Entzündungshemmung durch orale Steroide nach der Operation erforderlich. Präoperativ sollte mittels Bindehautsackkultur auf MRSA oder MRSE getestet und gegebenenfalls eine prophylaktische antibiotische Augentropfengabe erfolgen. Bei der Kataraktchirurgie wird empfohlen, die Inzision klein zu halten, die Augenoberfläche mit viskoelastischen Substanzen zu schützen und die Operation so kurz wie möglich zu gestalten.
QWas ist das Wichtigste in der Behandlung der akuten Phase?
A
Die ausreichende Entzündungshemmung der Augenoberfläche in der akuten Phase ist am wichtigsten. Neben einer systemischen Steroid-Pulstherapie (Methylprednisolon 500–1.000 mg/Tag für 3 Tage) 10) ist eine häufige lokale Gabe von Betamethason-Augentropfen erforderlich. Wenn die Hornhautepithelstammzellen in der akuten Phase erhalten bleiben, kann die Hornhauttransparenz wahrscheinlich aufrechterhalten werden. Ab Grad 2 kann eine frühzeitige Amnionmembrantransplantation auch bei schweren Fällen eine gute Sehprognose ermöglichen 8).
6. Pathophysiologie und detaillierter Entstehungsmechanismus
Die Pathophysiologie von SJS/TEN wird durch eine überschießende zelluläre Immunantwort auf Arzneimittelmetaboliten vor dem Hintergrund einer genetischen Prädisposition erklärt, gefolgt von ausgedehnter Epithelzellapoptose und sekundärer Entzündung und Gewebeschädigung. Obwohl die Zielstruktur (Epidermis und Schleimhautepithel) systemisch gleich ist, neigt die Entzündung an der Augenoberfläche aufgrund der Lage der Limbusstammzellen, der Durchblutung und der Zusammensetzung der Tränenflüssigkeit dazu, länger anzuhalten und direkt zum Verlust der Geweberegenerationsfähigkeit zu führen.
SJS/TEN werden als Typ-IV-Überempfindlichkeitsreaktion (verzögerter Typ) klassifiziert. Arzneimittelmetaboliten werden auf MHC-Klasse-I-Molekülen präsentiert, wobei CD8+ zytotoxische T-Lymphozyten (CTL) eine zentrale Rolle spielen 5). Aktivierte CD8+ T-Zellen sezernieren TNF-α und IFN-γ, was Keratinozyten zur Produktion von Stickstoffmonoxid (NO) anregt. Dieses NO fördert den Tod von Keratinozyten über den Fas/Fas-Ligand-Signalweg 5). Die weit verbreitete Apoptose von Keratinozyten führt zu Nekrose und Ablösung der gesamten Epidermis, was histopathologisch als vollständige Nekrose der Basalschicht der Epidermis beobachtet wird, einem charakteristischen Merkmal von SJS/TEN. Der Nachweis eines positiven Nikolsky-Zeichens und einer Nekrose der Basalschicht in einer Hautbiopsie ist hilfreich für die Abgrenzung von SSSS und anderen Erkrankungen.
Granulysin wurde als Hauptmediator des Keratinozyten-Todes bei SJS/TEN identifiziert 5). NK-Zellen vermitteln ebenfalls den Keratinozyten-Tod, indem der CD94/NKG2C-Rezeptor an HLA-E-Moleküle auf Keratinozyten bindet 5). TNF-α reguliert die Expression von Zelltod-assoziierten Molekülen in der Epidermis hoch, was letztlich zu einer ausgedehnten epidermalen Ablösung führt 5).
Darüber hinaus wird angenommen, dass eine Vaskulitis durch Ablagerung von Immunkomplexen in den subepithelialen Gefäßwänden der Haut und Schleimhäute beteiligt ist, und Mikrozirkulationsstörungen tragen zur Erosionsbildung und verzögerten Wundheilung bei. Die Hemmung der Prostaglandinsynthese, ein gemeinsamer Mechanismus von NSAIDs, wird ebenfalls als möglicher Faktor für die Entstehung von SJS/TEN vermutet 13).
Am Auge können bereits vor der Infiltration von Immunzellen Epithelablösungen und ein Verlust von Hemidesmosomen auftreten 8). In frühen Keratinozyten wird eine Vakuolisierung der Basalschicht beobachtet, obwohl keine Immunzellen vorhanden sind, was auf eine Zytokin-Dysregulation vor der Immuninfiltration hindeutet 8). Dies legt nahe, dass die Entzündung der Augenoberfläche der systemischen Arzneimittelreaktion vorausgeht oder gleichzeitig verläuft, und untermauert die Bedeutung der häufigen lokalen Steroidapplikation in der Akutphase nicht nur als symptomatische Therapie, sondern als krankheitsmodifizierender Eingriff.
In der Spätphase infiltrieren CD8+ zytotoxische T-Lymphozyten und greifen Keratinozyten an 8). In der chronischen Phase persistieren Neutrophile im Bindehautgewebe, was zu einer Dysregulation des Immunsystems führt und möglicherweise eine Schädigung der limbalen Stammzellen verursacht 8). Die anhaltende chronische Entzündung fördert die Fibrose und Narbenbildung der Bindehaut, was über eine Verminderung der Becherzellen, eine Verstopfung der akzessorischen Tränendrüsen und eine Meibom-Drüsen-Dysfunktion zu einer dauerhaften Störung des Tränenfilms führen kann. Um diese Kaskade in der chronischen Phase zu durchbrechen, wird manchmal eine langfristige Unterdrückung selbst leichter Entzündungen mit niedrig dosierten Steroid-Augentropfen durchgeführt.
Anatomie der kornealen epithelialen Stammzellen und Mechanismus ihres Verlusts
Korneale Epithelstammzellen befinden sich in den Basalzellen des Limbusepithels und machen weniger als 1% aller Basalzellen aus. Diese Stammzellen wandern zentripetal zur Hornhautmitte, während sie proliferieren und differenzieren, und erhalten so den gesamten Umsatz des Hornhautepithels. Ein anatomisches Merkmal des Limbusepithels sind die radiären Faltenstrukturen, die als Palisaden von Vogt (POV) bezeichnet werden und eine reiche Versorgung mit Blutgefäßen und Nerven sowie eine spezielle Mikroumgebung bilden, die die Stammzellnische darstellt. Die POV sind normalerweise oben und unten gut zu beobachten, können jedoch bei gesunden Augen unter 10 Jahren oder über 70 Jahren nicht deutlich sichtbar sein. Daher wird die Diagnose nicht allein anhand des Verschwindens der POV gestellt, sondern in Kombination mit Befunden wie konjunktivaler Epithelinvasion oder Unterschieden in der Epithelfärbung.
Wenn in der akuten Phase ein ausgedehnter kornealer und konjunktivaler Epitheldefekt auftritt und die Limbusstammzellen verloren gehen, bedeckt konjunktivales Epithel die Hornhautoberfläche, was zu Trübung und Vaskularisation führt. Dieser Prozess wird als Limbusstammzellinsuffizienz (limbal stem cell deficiency; LSCD) bezeichnet 8). SJS ist eine der Hauptursachen für chronische LSCD; in einer Studie mit 738 Augen einer einzelnen Einrichtung waren 10,4% auf SJS zurückzuführen 8). In den letzten Jahren wurden Versuche unternommen, den Schweregrad und die Verteilung der LSCD objektiv zu diagnostizieren, indem Methoden wie der Nachweis von Becherzellen mittels Impressionzytologie, die direkte Beobachtung von POV-Basalzellen mittels In-vivo-Konfokalmikroskopie und die schichtweise Bewertung der Epitheldicke mittels optischer Kohärenztomographie des vorderen Augenabschnitts kombiniert werden 8).
HLA-A*02:06 ist stark mit SJS/TEN mit schweren Augenkomplikationen bei Japanern assoziiert 12). Für verschiedene Medikamente wurden empfängliche HLA-Allele identifiziert, wie HLA-B*58:01 (Allopurinol), HLA-B*15:02 (Carbamazepin) und HLA-B*57:01 (Abacavir), und die Prävention durch Screening vor der Medikamentengabe wird in immer mehr Bereichen möglich 8). Diese HLA-Polymorphismen fungieren als Schlüsselmoleküle, die die Interaktion zwischen T-Zell-Rezeptor (TCR) und dem Medikament/Arzneimittelmetaboliten bestimmen. Es wurden die Hapten-Hypothese und die p-i-Hypothese (pharmakologische Interaktion) vorgeschlagen, wonach das Arzneimittelmolekül selbst oder sein reaktiver Metabolit direkt an die Peptidbindungsfurche des HLA-Moleküls bindet und die Präsentation von Selbstpeptiden verändert, wodurch eine abnormale autoreaktive T-Zell-Antwort ausgelöst wird. Die Aufklärung dieser molekularen Mechanismen bildet die Grundlage für zukünftige Präventionsstrategien und die sichere Auswahl von Medikamenten.
Bei PD-1-Inhibitor-induziertem SJS/TEN ist die Expression von PD-L1, die in normaler Haut normalerweise nicht nachweisbar ist, in Lymphozyten und Keratinozyten signifikant hochreguliert 5). Dies führt zum Tod von Keratinozyten durch aktivierte CD8+ T-Zellen 5).
QWarum bleiben dauerhafte Schäden an der Hornhaut zurück?
A
Wenn die epithelialen Stammzellen der Hornhaut (in den Basalzellen des Limbus) in der akuten Phase verloren gehen, wird eine Regeneration des Hornhautepithels unmöglich. Die Hornhautoberfläche wird von Bindehautgewebe mit Blutgefäßen und Bindegewebe bedeckt, undurchsichtig und unregelmäßig. In der chronischen Phase treibt das anhaltende Vorhandensein von Neutrophilen die Immunfehlregulation an und erhält die Stammzellschädigung aufrecht 8). Hinzu kommt eine Tränensekretionsinsuffizienz durch Verstopfung der Tränendrüsengänge, sodass trockene Augen und Hornhauttrübungen lebenslang bestehen bleiben.
Mit der zunehmenden Anwendung von ICIs steigt die Zahl der SJS/TEN-Berichte. In einer Untersuchung von 13 Fällen (China) mit Tislelizumab-induziertem SJS/TEN waren 9 männlich, das Durchschnittsalter betrug 73,15 ± 7,13 Jahre 5). Die Behandlungsschemata waren vielfältig, darunter Steroide allein, Steroide + IVIG, Steroide + IVIG + Cyclosporin, und 12 Patienten verbesserten sich 5).
Bei einem Fall von Tislelizumab-induziertem SJS/TEN, der auf die initiale Behandlung mit Steroiden und IVIG nicht ansprach, wurde eine Kombination aus TNF-α-Inhibitor (rekombinantes humanes TNF-Rezeptor-II-Antikörper-Fusionsprotein) und Blutreinigung eingesetzt, was zu einer Besserung führte 5). Die Anwendung von TNF-α-Inhibitoren zur Behandlung von SJS/TEN wird als neue Therapiestrategie beachtet.
Kultivierte Hornhautepitheltransplantate und regenerative Medizin
Die regenerative Medizin für Limbusstammzellinsuffizienz hat in den letzten Jahren große Fortschritte gemacht. Durch die Zugabe von Retinol und EGF zum Kulturmedium können Hornhautepitheltransplantate ohne Feederzellen und Serum hergestellt werden, und Transplantationsmaterialien ohne xenogene Substanzen werden klinisch eingesetzt 13). Die Transplantation von autologem Mundschleimhautepithel kann auch bei bilateraler LSCD als autologe Zellquelle genutzt werden und wird bei schweren Augenerkrankungen wie SJS angewendet 13). SLET verbreitet sich als einfacheres und kostengünstigeres Verfahren unter den LSCT-Methoden, und eine Übersicht bestehender Studien zeigt bessere Ergebnisse als CLET 9). Darüber hinaus ist ein Produkt, bei dem Mundschleimhautepithelzelltransplantate auf eine Amnionmatrix aufgebracht sind, als regeneratives Medizinprodukt zugelassen, und die klinische Anwendung bei bilateraler LSCD schreitet voran. Klinische Studien zu iPS-Zell-abgeleiteten Hornhautepithelzelltransplantaten werden auch in Japan durchgeführt, und sie werden als nächste Generation der Behandlung für Patienten mit bilateralen Erkrankungen erwartet, bei denen keine autologen Zellquellen verfügbar sind.
Es wurde berichtet, dass auch Sulfonamid-haltige Augentropfen systemisches SJS/TEN verursachen können. Sechs Tage nach Beginn der Brinzolamid-Augentropfen trat eine systemische Reaktion auf 99% der Körperoberfläche auf 1). Es wurde gezeigt, dass der Absorptionsweg über die Bindehaut und Nasenschleimhaut bei genetisch prädisponierten Individuen eine systemische Immunantwort auslösen kann 1). Diese Erkenntnis zeigt, dass selbst in der Augenheilkunde verwendete Augentropfen Auslöser schwerer systemischer Arzneimittelreaktionen sein können, was bei der Verschreibung an Patienten mit Arzneimittelallergien in der Vorgeschichte eine sorgfältige Abwägung erfordert.
Systemische Komplikationen im Zusammenhang mit SJS/TEN
Es wurden Fälle berichtet, bei denen SJS/TEN mit fulminantem Typ-1-Diabetes einherging 3). Es wird angenommen, dass eine systemische Immunantwort die Betazellen der Bauchspeicheldrüse zerstört, was die Bedeutung der Blutzuckerüberwachung während des SJS/TEN-Managements unterstreicht 3). Darüber hinaus wurden als systemische Komplikationen von SJS/TEN unter anderem akute interstitielle Pneumonie, akute Nierenschädigung, Leberfunktionsstörungen und Gerinnungsstörungen berichtet, sodass bei der systemischen Behandlung eine kontinuierliche Überwachung mehrerer Organe unerlässlich ist.
Patienten mit schweren okulären Spätfolgen von SJS/TEN benötigen lebenslang eine Kombination mehrerer Behandlungen wie Hornhauttransplantation, Limbusstammzelltransplantation, künstliche Hornhaut und Sklerallinsen. Darüber hinaus sind eine kontinuierliche Behandlung von trockenem Auge, Trichiasis und chronischer Konjunktivitis sowie regelmäßige Gesichtsfeld- und Augeninnendruckkontrollen und die Behandlung von sekundärem Glaukom und Katarakt erforderlich. Daher ist die Betreuung durch eine einzelne Einrichtung oder Abteilung schwierig, und ein langfristiges Nachsorgesystem unter der Leitung eines Hornhautspezialisten sowie eine multidisziplinäre Zusammenarbeit mit Dermatologie, Rheumatologie, Mund-Kiefer-Gesichtschirurgie und Rehabilitation sind unerlässlich. Für die Aufrechterhaltung der Lebensqualität der Patienten sind auch Sehbehindertenbetreuung und Arbeitsunterstützung wichtig, und eine frühzeitige Einführung der visuellen Rehabilitation wird empfohlen. Was das System der Kostenübernahme für medizinische Behandlungen betrifft, können schwere okuläre Spätfolgen von SJS/TEN teilweise im Rahmen der designierten seltenen Krankheit „Schweres Erythema exsudativum multiforme (akute Phase)“ und als Hornhauterkrankung als Folgeerkrankung abgedeckt werden, daher ist es wünschenswert, in Zusammenarbeit mit medizinischen Sozialarbeitern Informationen bereitzustellen.
Lu H, Xu W, Wu Y, Zhang M, Ma S. Ocular administration of brinzolamide leading to Stevens-Johnson syndrome/toxic epidermal necrolysis overlap: A case report and review. Medicine. 2025;104(49):e46362. doi:10.1097/MD.0000000000046362. PMID:41366987; PMCID:PMC12689009.
Jessica J. Padniewski, Erick Jacobson‐Dunlop, Sam Albadri, Sara Hylwa. Stevens–Johnson syndrome precipitated by Moderna Inc. COVID‐19 vaccine: a case‐based review of literature comparing vaccine and drug‐induced Stevens–Johnson syndrome/toxic epidermal necrolysis. Int J Dermatology. 2022;61(8):923-929. doi:10.1111/ijd.16222.
Zhang X, Huang D, Lou D, Si X, Mao J. Stevens-Johnson Syndrome/Toxic epidermal necrolysis complicated with fulminant type 1 diabetes mellitus: a case report and literature review. BMC endocrine disorders. 2024;24(1):172. doi:10.1186/s12902-024-01683-5. PMID:39218880; PMCID:PMC11367887.
Zhou Y, Xue H, Lu C, Zhang Y, Wu Q, Zhang J, et al. Treatment of Tislelizumab-Induced Toxic Epidermal Necrolysis and Agranulocytosis: A Case Report and Literature Review. Current drug safety. 2025;20(3):361-365. doi:10.2174/0115748863297885240604111018. PMID:38910480; PMCID:PMC12307955.
Yu H, Li Y, Qu X, Zhu J, Liu Z, Mu Z. Stevens-Johnson syndrome/toxic epidermal necrolysis induced by tislelizumab: a case report and literature review. Frontiers in immunology. 2025;16:1689877. doi:10.3389/fimmu.2025.1689877. PMID:41346629; PMCID:PMC12672898.
Zhang M, Wu R, Jia M, Sun S, Zhang L, Tang T. Sintilimab-induced erythema multiforme drug eruption in the treatment of sigmoid colon cancer: A case report and literature review. Medicine. 2023;102(41):e35659. doi:10.1097/MD.0000000000035659. PMID:37832081; PMCID:PMC10578730.
Zhang L, Yang P, Zhu Y, Liu K, Sun Z. Toxic epidermal necrolysis following lamotrigine replacement therapy in a woman planning pregnancy: a case report and literature review. BMC Womens Health. 2025;25:371. doi:10.1186/s12905-025-03796-y.
Jennifer C. W. Hu, Danielle Trief. A narrative review of limbal stem cell deficiency & severe ocular surface disease. Ann Eye Sci. 2023;8:13-13. doi:10.21037/aes-22-35.
Shanbhag SS, Nikpoor N, Rao Donthineni P, Singh V, Chodosh J, Basu S. Autologous limbal stem cell transplantation: a systematic review of clinical outcomes with different surgical techniques. The British journal of ophthalmology. 2020;104(2):247-253. doi:10.1136/bjophthalmol-2019-314081. PMID:31118185.
Sotozono C, Ueta M, Nakatani E, Kitami A, Watanabe H, Sueki H, et al. Predictive Factors Associated With Acute Ocular Involvement in Stevens-Johnson Syndrome and Toxic Epidermal Necrolysis. American journal of ophthalmology. 2015;160(2):228-237.e2. doi:10.1016/j.ajo.2015.05.002. PMID:25979679.
Ueta M, Tokunaga K, Sotozono C, Inatomi T, Yabe T, Matsushita M, Mitsuishi Y, Kinoshita S.. HLA class I and II gene polymorphisms in Stevens-Johnson syndrome with ocular complications in Japanese. Mol Vis. 2008;14:550-555. PMID:18385790; PMCID:PMC2274925.
Kinoshita S, Koizumi N, Ueta M, Sotozono C. New surgical approaches to the management of Stevens-Johnson syndrome and toxic epidermal necrolysis. Cornea. 2015;34 Suppl 11:S97-S103.
Kopieren Sie den Artikeltext und fügen Sie ihn in den KI-Assistenten Ihrer Wahl ein.
Artikel in die Zwischenablage kopiert
Öffnen Sie unten einen KI-Assistenten und fügen Sie den kopierten Text in den Chat ein.