ARS type 1
Gen gây bệnh: PITX2 (4q25)
Các bất thường chính: Bất thường đoạn trước mắt, bất thường răng, da thừa quanh rốn/thoát vị rốn, bất thường sọ mặt, bất thường tim mạch

Hội chứng Axenfeld-Rieger (ARS) là một nhóm bệnh bẩm sinh kết hợp các bất thường đoạn trước mắt và bất thường toàn thân. Nguyên nhân cơ bản là do rối loạn di chuyển và biệt hóa của tế bào mào thần kinh. Vào cuối thai kỳ, quá trình biến mất của các tế bào nội mô chưa biệt hóa bao phủ tiền phòng từ mống mắt và góc tiền phòng bị rối loạn, và phần còn lại của chúng gây ra sự hình thành các dải hoặc bám cao của mống mắt.
Bối cảnh lịch sử: Năm 1920, Axenfeld mô tả vòng Schwalbe sau (di lệch ra trước và dày lên của đường Schwalbe) và các mấu mống mắt. Năm 1934-1935, Rieger báo cáo thêm về giảm sản mống mắt, lệch đồng tử và đa đồng tử. Hiện nay được phân loại thành ba giai đoạn:
Tất cả những điều này được gọi chung là hội chứng Axenfeld-Rieger. 50-60% kèm glôcôm, di truyền trội trên nhiễm sắc thể thường và thường hai bên. Thường kết hợp với đục thủy tinh thể và lệch thể thủy tinh.
Dịch tễ học: Tỷ lệ hiện mắc trước đây được ước tính khoảng 1/200.000, nhưng các báo cáo gần đây ước tính từ 1/50.000 đến 1/100.0002)4). Không có khác biệt về giới tính, và thường được chẩn đoán ở giai đoạn trẻ sơ sinh hoặc trẻ nhỏ.
Phân loại di truyền như sau:
ARS type 1
Gen gây bệnh: PITX2 (4q25)
Các bất thường chính: Bất thường đoạn trước mắt, bất thường răng, da thừa quanh rốn/thoát vị rốn, bất thường sọ mặt, bất thường tim mạch
ARS type 2
Gen gây bệnh: 13q14 (chưa xác định)
Các bất thường chính: Bất thường đoạn trước mắt, glôcôm. Các bất thường toàn thân ít hơn type 1 và type 3
ARS type 3
Gen gây bệnh: FOXC1 (6p25)
Các bất thường chính: Bất thường đoạn trước mắt, glôcôm, mất thính lực thần kinh giác quan, thông liên nhĩ, bất thường thận, tổn thương chất trắng
Đột biến FOXC1 và PITX2 chiếm 40–70% các trường hợp ARS5). Tuy nhiên, ở 60% trường hợp ARS, gen gây bệnh vẫn chưa được xác định4), cho thấy tính đa dạng di truyền lớn.
Trong một phân tích từ cơ sở dữ liệu lớn về glôcôm khởi phát ở trẻ em và người trẻ, tỷ lệ chẩn đoán phân tử là 56,5%11). Đột biến FOXC1 chiếm 20,3%, PITX2 chiếm 17,4% và PAX6 chiếm 10,1%, và nhiều trường hợp không thể giải thích bằng các gen đã biết11).
Chúng được phân biệt dựa trên gen gây bệnh. Type 1 do đột biến PITX2 (4q25) và kèm theo bất thường răng, rốn và xương mặt. Type 3 do đột biến FOXC1 (6p25) và kèm theo mất thính lực, khuyết tật tim, bất thường thận và thần kinh. Type 2 nằm trên 13q14 nhưng gen gây bệnh chưa được xác định, với bất thường đoạn trước mắt và glôcôm là đặc điểm chính. Chẩn đoán có thể được xác nhận bằng xét nghiệm di truyền.
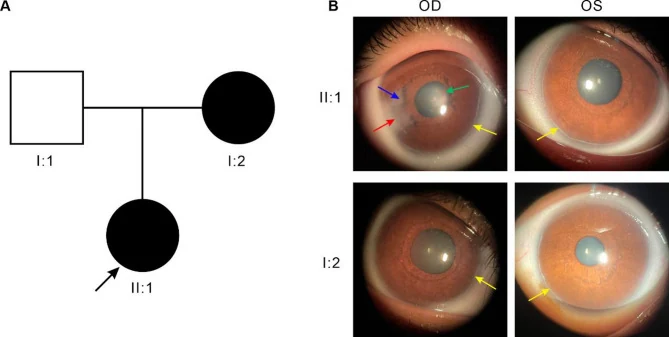
Các mục chính của dấu hiệu mắt được trình bày dưới đây.
| Dấu hiệu mắt | Đặc điểm |
|---|---|
| Vòng phôi sau | Đường Schwalbe lệch ra trước và dày lên |
| Mấu mống mắt | Dạng sợi mảnh đến dải rộng |
| Lệch đồng tử | Lệch về phía đối diện với vòng phôi thai sau |
| Giả đa đồng tử | Hình dạng như thủng ở mô đệm mống mắt |
| Lộn màng bồ đào | Lộn biểu mô sắc tố mống mắt |
Vòng phôi thai sau là tế bào chưa biệt hóa còn sót lại ở đường Schwalbe, xuất hiện dạng đường dọc theo rìa giác mạc, cách rìa 0,5–2,0 mm về phía trung tâm. Thường chỉ giới hạn một phần, không toàn bộ chu vi. Nếu đường Schwalbe nổi rõ dính vào mống mắt, gọi là bất thường Axenfeld; nếu kèm teo mô đệm mống mắt, gọi là bất thường Rieger.
Giác mạc thường trong suốt với cấu trúc nội mô bình thường, nhưng tiếp xúc vật lý với mô tồn dư có thể gây đục giác mạc thứ phát. Đục thường khu trú ở vùng ngoại vi và thường không ảnh hưởng trực tiếp đến thị lực. Tuy nhiên, ở đột biến FOXC1, đục và tân mạch giác mạc rõ rệt hơn, mức độ bất thường giác mạc lớn hơn và tần suất glôcôm cao hơn so với đột biến PITX21).
Dấu hiệu góc tiền phòng bao gồm bám mống mắt cao, di tích màng bồ đào dạng dây, và dày đường Schwalbe (vòng phôi thai sau). Cũng có báo cáo về trường hợp kèm vi cầu thể thủy tinh hoặc bán trật thể thủy tinh7).
Glôcôm xảy ra ở 50–60% trường hợp. Tăng nhãn áp có thể xảy ra từ giai đoạn sơ sinh, nhưng hầu hết biểu hiện ở thời thơ ấu đến thanh niên. Một số trường hợp được chẩn đoán sau khi thị lực giảm dần, do đó cần không bỏ sót các dấu hiệu glôcôm và thay đổi ở đoạn trước6).
Trong trường hợp của Li et al. (2021) ở một bé trai 7 tuổi (ARS type 3, đột biến de novo FOXC1), đường kính giác mạc 14 mm, chiều dài trục 27,16/26,56 mm, tỷ lệ C/D 0,9, nhãn áp 33/20 mmHg. Cả hai mắt cần phẫu thuật chống glôcôm từ ngày thứ 36 sau sinh5).
Dấu hiệu toàn thân như sau:
Khoảng 50-60% bệnh nhân ARS bị glôcôm. Thường khởi phát ở thời thơ ấu đến đầu tuổi trưởng thành, nhưng cũng có trường hợp tăng nhãn áp từ giai đoạn trẻ sơ sinh. Cần đo nhãn áp thường xuyên và đánh giá thần kinh thị giác. Để biết chi tiết, hãy tham khảo phần “Phương pháp điều trị tiêu chuẩn”.
ARS di truyền trội trên nhiễm sắc thể thường với độ thâm nhập hoàn toàn. Tuy nhiên, ngay cả trong cùng một gia đình mang cùng một đột biến gen, vẫn có sự khác biệt lớn về biểu hiện lâm sàng (tính biểu hiện thay đổi)1).
Trong một nghiên cứu thuần tập lớn, đột biến FOXC1 và PITX2 có liên quan đến phổ glôcôm rộng từ thời thơ ấu đến tuổi trưởng thành9). Các trường hợp được chẩn đoán lâm sàng ban đầu là glôcôm bẩm sinh nguyên phát (PCG) đôi khi được phân loại lại thông qua xét nghiệm di truyền, và xét nghiệm di truyền góp phần chẩn đoán chính xác type khi các dấu hiệu ở đoạn trước của mắt ở trẻ sơ sinh còn tinh tế.
Trong các trường hợp có vi mất đoạn quanh gen PITX2, sự chồng lấn của các mất đoạn NEUROG2, UGT8 và NDST4 có thể gây chậm phát triển và khuyết tật trí tuệ 8)3).
Kawanami et al. (2023) báo cáo một bé trai người Nhật 3 tuổi có vi mất đoạn 2,5 Mb tại 4q25 (bao gồm PITX2, NEUROG2 và ANK2). Bé có thoát vị rốn, u mống mắt và chậm phát triển, nhưng điện tâm đồ bình thường mặc dù mất đoạn ANK2. Sự thiếu hụt một bản sao của NEUROG2 được cho là nguyên nhân tiềm năng gây chậm phát triển 8).
Vì là bệnh trội trên nhiễm sắc thể thường, xác suất di truyền đột biến từ cha mẹ là 50%. Tính thâm nhập hoàn toàn nhưng kiểu hình thay đổi, và mức độ nghiêm trọng của triệu chứng có thể khác nhau đáng kể ngay cả với cùng một đột biến 1). Khuyến cáo thực hiện xét nghiệm di truyền và tư vấn di truyền.
Chẩn đoán ARS dựa trên bất thường góc tiền phòng và mống mắt ở cả hai mắt. Sự hiện diện của vòng phôi sau (posterior embryotoxon) với sự bám dính một phần của mống mắt xung quanh là điều kiện để chẩn đoán 13). Nếu không thấy vòng phôi sau bằng đèn khe, cần soi góc tiền phòng. Nếu có bất thường toàn thân, bệnh nhân được chuyển đến bác sĩ nhi khoa để đánh giá toàn diện như một phần của hội chứng ARS 13).
Lưu ý rằng 8-15% dân số bình thường có vòng phôi sau nhẹ, nhưng riêng điều này không đi kèm với glôcôm hay các bệnh khác. Tiền sử gia đình cũng quan trọng trong chẩn đoán.
Trong hệ thống phân loại glôcôm trẻ em theo Hướng dẫn Điều trị Glôcôm (phiên bản thứ 5), ARS được phân loại là một ví dụ điển hình của glôcôm liên quan đến bất thường phát triển mắt bẩm sinh 13). Chẩn đoán được đưa ra khi các bất thường mắt tồn tại từ khi sinh đáp ứng tiêu chuẩn chẩn đoán glôcôm trẻ em.
Dưới đây là các bệnh chính cần phân biệt với ARS.
| Bệnh | Khác biệt với ARS |
|---|---|
| Hội chứng ICE | Một bên, mắc phải, ưu thế nữ |
| Bất thường Peters | Đục giác mạc trung tâm, khiếm khuyết màng Descemet |
| Vô mống mắt | Pannus giác mạc, giảm sản hố trung tâm |
| Loạn dưỡng giác mạc đa hình sau | Hai bên, gia đình, không khác biệt giới tính |
Hội chứng ICE (như teo mống mắt tiến triển, hội chứng Chandler) cần được phân biệt với ARS, nhưng điểm khác biệt chính là ICE một bên và mắc phải, trong khi ARS hai bên và bẩm sinh.
Hiện không có phương pháp điều trị triệt căn cho ARS, và việc quản lý glôcôm cùng theo dõi các biến chứng toàn thân là trọng tâm điều trị. Phác đồ điều trị tuân theo glôcôm phát triển sớm (glôcôm bẩm sinh nguyên phát: PCG) 13).
Glôcôm xảy ra ở khoảng 50-60% các trường hợp ARS. Điều trị bằng thuốc tuân theo hướng dẫn chung về glôcôm, nhưng thường không hiệu quả.
Thuốc ức chế sản xuất thủy dịch
Thuốc chẹn beta: Một trong những lựa chọn đầu tay. An toàn và hiệu quả nhưng thường không hiệu quả ở trẻ em.
Thuốc nhỏ ức chế men carbonic anhydrase (CAI): như brinzolamide. Có thể kết hợp với thuốc chẹn beta.
Thuốc chủ vận alpha-2 (brimonidine): Chống chỉ định ở trẻ dưới 2 tuổi do tác dụng thần kinh tâm thần (ngưng thở, nhịp tim chậm, hạ huyết áp, giảm trương lực cơ, ức chế thần kinh trung ương) 13).
Thuốc tăng cường dẫn lưu thủy dịch
Các chất tương tự prostaglandin: như latanoprost, travoprost. Hiệu quả ở trẻ em yếu hơn so với người lớn 13).
Ví dụ: Ở một bé trai 7 tuổi, quản lý dài hạn bằng travoprost + brinzolamide 5). Ở một nam giới 77 tuổi, nhãn áp 35 mmHg dù dùng latanoprost/timolol + brinzolamide, cho thấy khó kiểm soát 2).
Có báo cáo rằng không có sự khác biệt về hiệu quả giữa thuốc chủ vận thụ thể FP prostanoid và thuốc chẹn beta 13).
Ở trẻ sơ sinh và trẻ nhỏ, liều thuốc nhỏ mắt tương đối lớn so với cân nặng và diện tích bề mặt cơ thể, do đó nên sử dụng nồng độ thuốc thấp nhất có thể 13).
Nếu không kiểm soát được nhãn áp bằng thuốc, phẫu thuật sẽ được thực hiện10)13).
Các biến chứng sau GDD bao gồm: tiền phòng nông 13,6%, nhãn áp thấp 11,7%, tràn dịch hắc mạc 8,3% và viêm nội nhãn 1,7%14).
Chakraborty và cộng sự (2022) báo cáo một trường hợp bong võng mạc liên quan đến ARS (nam 15 tuổi). Kèm theo vi thể thủy tinh thể và bán trật thể thủy tinh, sau cắt dịch kính nhãn áp tăng lên 41 mmHg và hình thành u nhú. Đã thực hiện quang đông thể mi diode và cuối cùng nhãn áp đạt 18 mmHg7).
Tỷ lệ thành công của phẫu thuật góc thấp hơn so với PCG 13). Trong phẫu thuật cắt bè củng mạc kết hợp MMC, tỷ lệ thành công lâu dài 2 năm khoảng 59%; với thiết bị dẫn lưu tăng nhãn áp, tỷ lệ được báo cáo là 87% ở 12 tháng và 77% ở 24 tháng 14). Các trường hợp kháng trị có thể cần phẫu thuật nhiều lần.
Nguyên nhân cơ bản của ARS là khiếm khuyết trong di chuyển và biệt hóa của tế bào mào thần kinh. Sự phát triển bị suy giảm của tế bào mào thần kinh ở tiền phòng, góc tiền phòng, xương mặt, răng, hệ tim mạch và da quanh rốn dẫn đến dị dạng đa cơ quan.
Vào cuối thai kỳ, các tế bào nội mô chưa biệt hóa lót tiền phòng thường biến mất khỏi mống mắt và góc. Trong ARS, quá trình biến mất này bị suy giảm, và các tế bào nội mô chưa biệt hóa tồn tại trên mống mắt, gây ra sự hình thành các dải. Ở góc, xảy ra bám cao của mống mắt, che phủ cơ học bè củng mạc.
Về mô học, có một lớp đơn tế bào giống nội mô với màng giống màng Descemet kéo dài bất thường từ bề mặt sau giác mạc qua tiền phòng, góc và bề mặt mống mắt. Màng này hiện diện ở các góc phần tư có lộn màng bồ đào và lệch đồng tử, trong khi teo mống mắt được quan sát thấy ở các góc phần tư đối diện.
FOXC1 và PITX2 đều là yếu tố phiên mã, liên kết với các trình tự DNA cụ thể và điều hòa biểu hiện gen hạ nguồn. Cả hai hoạt động hiệp đồng trong sự phát triển của đoạn trước mắt và điều hòa các gen đích chung hạ nguồn 3). Miền forkhead của FOXC1 (miền liên kết DNA gồm 110 axit amin) là quan trọng nhất về mặt chức năng 2), và các đột biến trong miền này được cho là có liên quan chặt chẽ hơn đến các triệu chứng tâm thần kinh.
Hai cơ chế làm tăng nhãn áp đã được xác định:
Mức độ khuyết tật mống mắt và số lượng mấu mống mắt ở góc không nhất thiết tương quan với mức độ nghiêm trọng của bệnh tăng nhãn áp. Tuy nhiên, dính mống mắt góc nặng làm tăng nguy cơ mắc bệnh tăng nhãn áp.
Đột biến FOXC1 thúc đẩy bệnh tăng nhãn áp bẩm sinh nhiều hơn so với các đột biến khác 1), và các bất thường về hình thái của thể mi và góc thoát nước có thể góp phần làm tăng nhãn áp 1).
Ở chuột mang đột biến FOXC1, quan sát thấy sự giảm sợi collagen ở nhu mô giác mạc, bất thường cấu trúc và tổn thương tế bào nhu mô 1). Hơn nữa, FOXC1 hoạt động như một yếu tố ức chế sự hình thành mạch máu giác mạc (thông qua kiểm soát khả dụng sinh học của VEGF) 1), và sự mất ức chế này do đột biến FOXC1 dẫn đến hình thành mạch máu giác mạc.
FOXC1, như một yếu tố phiên mã họ FOX, cũng đóng vai trò quan trọng trong sự phát triển của não 4).
Trong một tổng quan hệ thống, đã báo cáo rằng bất thường chất trắng xuất hiện ở 41,3% các trường hợp ARS 4). Đột biến FOXC1 có thể gây ra bệnh mạch máu nhỏ não, tăng tín hiệu chất trắng, giãn khoang quanh mạch, xuất huyết vi thể và nhồi máu ổ khuyết.
Ohkubo và cộng sự (2025) đã xác nhận tổn thương chất trắng quanh não thất, giãn khoang quanh mạch, và tình trạng xoắn vặn và giãn động mạch đốt sống thân nền trên MRI não của một bé trai Nhật Bản 2 tuổi (đột biến FOXC1: c.240del, p.Y81Ifs21). Cha của bé có tiền sử nhồi máu não ở tuổi 18 4).
Trong một tổng quan 95 trường hợp đột biến FOXC1, các triệu chứng tâm thần kinh (khó khăn học tập, động kinh, khuyết tật trí tuệ, hoang tưởng ghen tuông, v.v.) được tìm thấy ở 6,3%, và các triệu chứng tâm thần kinh xuất hiện ở 83,3% các trường hợp có đột biến vùng forkhead 2).
Có. Có một tổng quan hệ thống báo cáo rằng bất thường chất trắng xuất hiện ở khoảng 41% các trường hợp ARS có đột biến FOXC1 4). Đột biến FOXC1 có thể liên quan đến bệnh mạch máu nhỏ não và nguy cơ đột quỵ, và đặc biệt ở ARS type FOXC1, việc theo dõi thần kinh dài hạn là quan trọng.
Trong những năm gần đây, nhiều đột biến mới đã được báo cáo thông qua giải trình tự thế hệ mới và giải trình tự toàn bộ gen.
Wowra và cộng sự (2024) đã xác định một đoạn mất lớn (đột biến mới) bao gồm một phần exon 1 và toàn bộ vùng 3’UTR của FOXC1 ở ba chị em người Ba Lan mắc ARS. Kiểu hình rất khác nhau ngay trong cùng một gia đình, và ban đầu họ bị chẩn đoán nhầm là hội chứng Chandler 1).
Jiang và cộng sự (2024) đã xác định một sự sắp xếp lại bộ gen phức tạp bao gồm mất đoạn 6,15 Mb trên nhiễm sắc thể 4q25 chứa PITX2, đảo đoạn 45,71 Mb và mất 14 bp trong một gia đình người Trung Quốc mắc ARS type 1. Một bé gái 11 tuổi có nhãn áp 43,5/44,0 mmHg 3).
Các báo cáo đột biến mới khác bao gồm FOXC1 p.Phe136Leu (vùng forkhead) 2), FOXC1 p.S82R (đột biến de novo) 5), và FOXC1 c.240del, p.Y81Ifs21 4).
Yoshino và cộng sự (2024) đã báo cáo một trường hợp nam giới Nhật Bản 77 tuổi mắc ARS type 3. Hoang tưởng ghen tuông xuất hiện từ năm 72 tuổi, và bệnh não chất trắng đã được xác nhận. Trong một tổng quan y văn về 95 trường hợp đột biến FOXC1, các triệu chứng tâm thần kinh được tìm thấy ở 6,3% (6/95), và 83,3% (5/6) trong số đó có đột biến vùng forkhead 2).
Phát hiện này cho thấy vùng chức năng của đột biến FOXC1 có thể liên quan đến biểu hiện của các triệu chứng tâm thần kinh, nhấn mạnh tầm quan trọng của việc theo dõi lâu dài từ góc độ sức khỏe tâm thần.
Đột biến FOXC1 được cho là có thể gây ra CSVD và tăng nguy cơ đột quỵ 4). Phòng ngừa và can thiệp sớm bệnh mạch máu thần kinh ở bệnh nhân ARS là chủ đề nghiên cứu trong tương lai.
Sự hiểu biết về cơ chế bệnh sinh của “xơ cứng hóa” giác mạc do đột biến FOXC1 đang tiến triển 1). Sự hiểu biết về cơ chế phân tử của đục giác mạc được kỳ vọng sẽ dẫn đến sự phát triển các liệu pháp mới sử dụng liệu pháp gen, thuốc chống xơ hóa và vật liệu sinh học 1).