ARS type 1
Gène causal : PITX2 (4q25)
Anomalies principales : anomalies du segment antérieur de l’œil, anomalies dentaires, excès de peau péri-ombilicale / hernie ombilicale, anomalies craniofaciales, anomalies cardiovasculaires

Le syndrome d’Axenfeld-Rieger (SAR) est un groupe de maladies congénitales associant des anomalies du segment antérieur de l’œil et des anomalies systémiques. La cause fondamentale est une anomalie de la migration et de la différenciation des cellules de la crête neurale. À la fin de la vie fœtale, le processus normal de disparition des cellules endothéliales indifférenciées recouvrant la chambre antérieure, de l’iris et de l’angle, est perturbé, et leur persistance entraîne la formation de brides et une insertion haute de l’iris.
Contexte historique : En 1920, Axenfeld a décrit l’anneau embryonnaire postérieur (déplacement antérieur et épaississement de la ligne de Schwalbe) et les processus iriens. En 1934-1935, Rieger a ajouté l’hypoplasie de l’iris, la déviation pupillaire et la polycorie. Actuellement, la classification comprend trois stades :
L’ensemble est appelé syndrome d’Axenfeld-Rieger. Un glaucome survient dans 50 à 60 % des cas, la transmission est autosomique dominante et généralement bilatérale. Une cataracte et une ectopie du cristallin sont également fréquemment associées.
Épidémiologie : la prévalence était estimée à environ 1/200 000, mais des rapports récents l’estiment entre 1/50 000 et 1/100 0002)4). Il n’y a pas de différence de sexe et le diagnostic est souvent posé dans la petite enfance.
Classification génétique :
ARS type 1
Gène causal : PITX2 (4q25)
Anomalies principales : anomalies du segment antérieur de l’œil, anomalies dentaires, excès de peau péri-ombilicale / hernie ombilicale, anomalies craniofaciales, anomalies cardiovasculaires
ARS type 2
Gène causal : 13q14 (non confirmé)
Anomalies principales : anomalies du segment antérieur de l’œil, glaucome. Les anomalies systémiques sont moins fréquentes que dans les types 1 et 3.
ARS type 3
Gène causal : FOXC1 (6p25)
Anomalies principales : anomalies du segment antérieur de l’œil, glaucome, surdité neurosensorielle, communication interauriculaire, anomalies rénales, lésions de la substance blanche
Les mutations de FOXC1 et PITX2 représentent 40 à 70 % des cas d’ARS5). Cependant, dans 60 % des cas d’ARS, le gène causal n’est pas identifié4), ce qui indique une grande diversité génétique.
Une analyse d’un large registre de glaucome à début juvénile ou infantile a montré un taux de diagnostic moléculaire de 56,5 %11). Les mutations FOXC1 représentaient 20,3 %, PITX2 17,4 % et PAX6 10,1 %, et de nombreux cas ne peuvent être expliqués par les gènes connus11).
Ils se distinguent par le gène causal. Le type 1 est dû à une mutation de PITX2 (4q25) et s’accompagne d’anomalies dentaires, ombilicales et des os du visage. Le type 3 est dû à une mutation de FOXC1 (6p25) et s’accompagne de surdité, de malformations cardiaques, d’anomalies rénales et neurologiques. Le type 2 est localisé en 13q14 mais le gène causal n’est pas confirmé ; il se caractérise principalement par des anomalies du segment antérieur et un glaucome. Le diagnostic peut être confirmé par un test génétique.
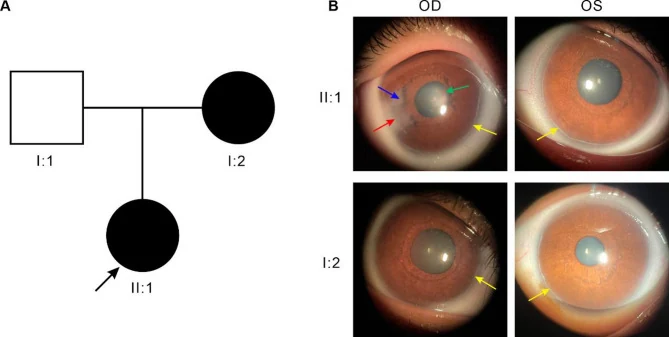
Les principaux signes oculaires sont présentés ci-dessous.
| Signe oculaire | Caractéristiques |
|---|---|
| Anneau embryonnaire postérieur | Déplacement antérieur et épaississement de la ligne de Schwalbe |
| Processus iriens | Fins à larges bandes |
| Déviation pupillaire | Déviation dans la direction opposée à l’anneau embryonnaire postérieur |
| Pseudopolycorie | Aspect perforé du stroma irien |
| Eversion uvéale | Rétroversion de l’épithélium pigmentaire irien |
L’anneau embryonnaire postérieur est une persistance de cellules indifférenciées au niveau de la ligne de Schwalbe, observée sous forme d’une ligne le long du limbe, décalée de 0,5 à 2,0 mm vers le centre. Elle est souvent partielle et non circonférentielle. Si la ligne de Schwalbe proéminente adhère à l’iris, on parle d’anomalie d’Axenfeld ; si elle s’accompagne d’une atrophie du stroma irien, on parle d’anomalie de Rieger.
La cornée est généralement transparente avec un endothélium normal, mais un contact physique avec le tissu résiduel peut entraîner une opacité cornéenne secondaire. L’opacité est souvent limitée à la périphérie et n’affecte généralement pas directement la vision. Cependant, dans les mutations FOXC1, l’opacité cornéenne et la néovascularisation sont plus marquées, et l’anomalie cornéenne est plus sévère qu’avec les mutations PITX2, avec une fréquence plus élevée de glaucome1).
Les signes angulaires comprennent une insertion haute de l’iris, des reliquats uvéaux en corde, et un épaississement de la ligne de Schwalbe (anneau embryonnaire postérieur). Des cas associant une microsphérophakie et une subluxation du cristallin ont également été rapportés7).
Le glaucome complique 50 à 60 % des cas. Une élévation de la pression intraoculaire peut survenir dès la petite enfance, mais elle se manifeste le plus souvent dans l’enfance ou chez le jeune adulte. Certains cas sont diagnostiqués à l’occasion d’une baisse progressive de l’acuité visuelle ; il est important de ne pas négliger les signes oculaires antérieurs et les modifications glaucomateuses6).
Li et al. (2021) ont rapporté le cas d’un garçon de 7 ans (ARS type 3, mutation de novo FOXC1) présentant un diamètre cornéen de 14 mm, une longueur axiale de 27,16/26,56 mm, un rapport C/D de 0,9 et une PIO de 33/20 mmHg. Une chirurgie antiglaucomateuse bilatérale a été nécessaire dès le 36e jour de vie5).
Les signes systémiques sont les suivants :
Environ 50 à 60 % des patients atteints d’ARS développent un glaucome. Il survient souvent pendant l’enfance ou le jeune âge adulte, mais peut aussi se manifester dès la petite enfance par une élévation de la pression intraoculaire. Une mesure régulière de la pression intraoculaire et une évaluation du nerf optique sont nécessaires. Pour plus de détails, voir la section « Traitement standard ».
L’ARS est une maladie autosomique dominante à pénétrance complète. Cependant, même au sein d’une même famille, des personnes portant la même mutation génétique peuvent présenter des tableaux cliniques très variables (expressivité variable)1).
Dans une étude de cohorte à grande échelle, les mutations de FOXC1 et PITX2 étaient associées à un large spectre de glaucome allant de l’enfance à l’âge adulte9). Des cas initialement diagnostiqués cliniquement comme un glaucome congénital primitif (PCG) peuvent être reclassés après test génétique. Lorsque les signes du segment antérieur chez le nourrisson sont subtils, le test génétique peut contribuer à un diagnostic précis du type de maladie.
Dans les cas présentant une microdélétion autour du gène PITX2, la délétion concomitante de NEUROG2, UGT8 et NDST4 peut entraîner un retard de développement et une déficience intellectuelle8)3).
Kawanami et al. (2023) ont rapporté le cas d’un garçon japonais de 3 ans présentant une microdélétion de 2,5 Mb en 4q25 (incluant PITX2, NEUROG2 et ANK2). Il présentait une hernie ombilicale, un colobome irien et un retard de développement, mais l’ECG était normal malgré la délétion d’ANK2. L’haploinsuffisance de NEUROG2 a été considérée comme une cause candidate du retard de développement8).
En raison du mode de transmission autosomique dominant, la probabilité de transmission d’un parent porteur de la mutation est de 50 %. La pénétrance est complète, mais le phénotype varie : la sévérité des symptômes peut différer considérablement pour une même mutation1). Un test génétique et un conseil génétique sont recommandés.
Le diagnostic de l’ARS repose sur des anomalies de l’angle et de l’iris bilatérales. La condition diagnostique est la présence d’un embryotoxon postérieur avec une adhérence partielle de l’iris périphérique13). Si l’embryotoxon postérieur n’est pas visible à la lampe à fente, une gonioscopie est nécessaire. En cas d’anomalies systémiques, un bilan complet en pédiatrie est demandé pour un syndrome ARS13).
À noter qu’un embryotoxon postérieur léger est observé chez 8 à 15 % de la population normale, mais il n’est pas associé à un glaucome en l’absence d’autres signes. L’interrogatoire des antécédents familiaux est également important pour le diagnostic.
Dans la classification du glaucome pédiatrique selon les lignes directrices pour le glaucome (5e édition), l’ARS est considérée comme un exemple typique de glaucome associé à une anomalie congénitale du développement oculaire13). Le diagnostic est posé lorsque des anomalies oculaires congénitales présentes à la naissance répondent aux critères du glaucome pédiatrique.
Les principales maladies à différencier de l’ARS sont présentées ci-dessous.
| Maladie | Différence avec l’ARS |
|---|---|
| Syndrome ICE | Unilatéral, acquis, prédominance féminine |
| Anomalie de Peters | Opacité cornéenne centrale, déficit de la membrane de Descemet |
| Aniridie | Pannus cornéen, hypoplasie fovéolaire |
| Dystrophie cornéenne polymorphe postérieure | Bilatéral, familial, pas de différence de sexe |
Le syndrome ICE (atrophie irienne progressive, syndrome de Chandler, etc.) nécessite une différenciation importante de l’ARS, mais le point de différenciation majeur est que l’ICE est unilatéral et acquis, tandis que l’ARS est bilatéral et congénital.
Il n’existe actuellement aucun traitement curatif du SAR lui-même. La prise en charge repose principalement sur le contrôle du glaucome et la surveillance des complications systémiques. La stratégie thérapeutique suit celle du glaucome congénital primitif (GCP) 13).
Le glaucome complique environ 50 à 60 % des cas de SAR. Le traitement médicamenteux suit celui du glaucome général, mais il est souvent inefficace.
Inhibiteurs de la production d'humeur aqueuse
Bêta-bloquants : l’un des traitements de première intention. Sûrs et efficaces, mais souvent inefficaces chez l’enfant.
Inhibiteurs de l’anhydrase carbonique (IAC) en collyre : brinzolamide, etc. Peuvent être associés aux bêta-bloquants.
Alpha-2 agonistes (brimonidine) : contre-indiqués avant l’âge de 2 ans en raison d’effets neuropsychiques (apnée, bradycardie, hypotension, hypotonie, dépression du SNC) 13).
Médicaments favorisant l'écoulement de l'humeur aqueuse
Analogues des prostaglandines : latanoprost, travoprost, etc. Leur efficacité chez l’enfant est considérée comme moindre que chez l’adulte 13).
Exemple : Un garçon de 7 ans a été contrôlé à long terme par travoprost + brinzolamide 5). Chez un homme de 77 ans, le latanoprost, le timolol et le brinzolamide n’ont pas permis de contrôler la PIO (35 mmHg) 2).
Une étude rapporte qu’il n’y a pas de différence d’efficacité entre les agonistes des récepteurs FP des prostanoides et les bêta-bloquants 13).
Chez les nourrissons, la dose de collyre est relativement élevée par rapport au poids et à la surface corporelle. Il convient donc d’utiliser des concentrations aussi faibles que possible 13).
Si le contrôle de la pression intraoculaire n’est pas obtenu par traitement médicamenteux, une intervention chirurgicale est réalisée 10)13).
Les complications postopératoires du GDD rapportées incluent : chambre antérieure peu profonde 13,6 %, hypotonie 11,7 %, décollement chorọidien 8,3 %, endophtalmie 1,7 % 14).
Chakraborty et al. (2022) ont rapporté un cas de décollement de rétine associé au syndrome d’Axenfeld-Rieger (garçon de 15 ans). Il présentait une microsphérophakie et une subluxation du cristallin ; après vitrectomie, la PIO a augmenté à 41 mmHg, formant un staphylome scléral. Une cyclophotocoagulation au diode a été réalisée, aboutissant à une PIO finale de 18 mmHg 7).
Le taux de succès de la chirurgie de l’angle est inférieur à celui du PCG 13). Pour la trabéculectomie avec MMC, le taux de succès à long terme à 2 ans est d’environ 59 % ; pour les GDD, il est de 87 % à 12 mois et de 77 % à 24 mois 14). Les cas réfractaires peuvent nécessiter plusieurs interventions chirurgicales.
La cause fondamentale de l’ARS est un défaut de migration et de différenciation des cellules de la crête neurale. Le développement altéré des cellules de la crête neurale dans la chambre antérieure, l’angle du segment antérieur, les os du visage, les dents, le système cardiovasculaire et la peau péri-ombilicale entraîne des malformations multi-organiques.
À la fin de la période fœtale, les cellules endothéliales indifférenciées recouvrant la chambre antérieure disparaissent normalement de l’iris et de l’angle. Dans l’ARS, ce processus de disparition est perturbé, laissant des cellules endothéliales indifférenciées résiduelles sur l’iris, formant des bandes. Dans l’angle, cela provoque une insertion haute de l’iris, recouvrant mécaniquement le trabéculum.
Histologiquement, on observe une extension anormale d’une monocouche de cellules de type endothélial avec une membrane de type descemétique depuis la face postérieure de la cornée jusqu’à la chambre antérieure, l’angle et la surface de l’iris. La membrane est présente dans les quadrants présentant une ectropion uvéal et une déviation pupillaire, tandis qu’une atrophie irienne est observée dans les quadrants opposés.
FOXC1 et PITX2 sont tous deux des facteurs de transcription qui se lient à des séquences d’ADN spécifiques pour réguler l’expression des gènes en aval. Ils agissent en synergie dans le développement du segment antérieur de l’œil et régulent des gènes cibles communs en aval 3). Le domaine forkhead de FOXC1 (domaine de liaison à l’ADN de 110 acides aminés) est le plus important fonctionnellement 2), et les mutations de ce domaine sont suggérées comme étant plus fortement associées aux symptômes neuropsychiatriques.
Deux mécanismes d’augmentation de la pression intraoculaire sont identifiés.
Le degré de l’absence d’iris et la quantité de processus irido-cornéens ne sont pas nécessairement corrélés à la sévérité du glaucome. Cependant, un degré élevé de synéchie irienne dans l’angle prédispose au glaucome.
Les mutations FOXC1 favorisent davantage le glaucome congénital que d’autres mutations 1), et les anomalies morphologiques du corps ciliaire et de l’angle de drainage peuvent contribuer à l’augmentation de la PIO 1).
Chez les souris mutantes FOXC1, on observe une diminution et des anomalies structurelles des fibres de collagène du stroma cornéen, ainsi qu’une altération des cellules du stroma cornéen 1). De plus, FOXC1 agit comme un inhibiteur de la néovascularisation cornéenne (via le contrôle de la biodisponibilité du VEGF) 1) ; la perte de cette inhibition due à la mutation FOXC1 entraîne une néovascularisation cornéenne.
FOXC1, en tant que facteur de transcription de la famille FOX, joue également un rôle important dans le développement cérébral 4).
Une revue systématique a rapporté que des anomalies de la substance blanche apparaissent dans 41,3 % des cas d’ARS 4). Les mutations FOXC1 peuvent induire une maladie des petits vaisseaux cérébraux (CSVD), des hypersignaux de la substance blanche, des espaces périvasculaires élargis, des microhémorragies et des infarctus lacunaires.
Ohkubo et al. (2025) ont confirmé, par IRM cérébrale, des lésions périventriculaires de la substance blanche, des espaces périvasculaires élargis et une tortuosité-dilatation de l’artère vertébrobasilaire chez un garçon japonais de 2 ans (mutation FOXC1 : c.240del, p.Y81Ifs21). Son père avait des antécédents d’infarctus cérébral à 18 ans 4).
Une revue de 95 cas de mutations FOXC1 a révélé des symptômes neuropsychiatriques (difficultés d’apprentissage, épilepsie, déficience intellectuelle, jalousie délirante, etc.) dans 6,3 % des cas, et 83,3 % des cas porteurs de mutations du domaine forkhead présentaient des symptômes neuropsychiatriques 2).
Oui. Une revue systématique indique que des anomalies de la substance blanche apparaissent dans environ 41 % des cas d’ARS avec mutation FOXC1 4). Les mutations FOXC1 pourraient être associées à un risque de maladie des petits vaisseaux cérébraux et d’accident vasculaire cérébral ; un suivi neurologique à long terme est donc important, en particulier dans l’ARS liée à FOXC1.
Ces dernières années, de nombreuses nouvelles mutations ont été rapportées grâce au séquençage de nouvelle génération et au séquençage du génome entier.
Wowra et al. (2024) ont identifié une grande délétion (nouvelle mutation) incluant une partie de l’exon 1 de FOXC1 et l’ensemble de la région 3’UTR dans l’ARS de trois sœurs polonaises. Les phénotypes étaient très différents au sein de la même famille, et un diagnostic erroné de syndrome de Chandler avait été posé initialement 1).
Jiang et al. (2024) ont identifié un réarrangement génomique complexe comprenant une délétion de 6,15 Mb du chromosome 4q25 contenant PITX2, une inversion de 45,71 Mb et une délétion de 14 pb dans une famille chinoise atteinte d’ARS de type 1. Une fillette de 11 ans présentait une PIO de 43,5/44,0 mmHg 3).
Parmi les autres nouvelles mutations rapportées figurent FOXC1 p.Phe136Leu (domaine forkhead) 2), FOXC1 p.S82R (mutation de novo) 5), et FOXC1 c.240del, p.Y81Ifs21 4).
Yoshino et al. (2024) ont rapporté le cas d’un homme japonais de 77 ans atteint d’ARS de type 3. Des idées délirantes de jalousie sont apparues à 72 ans, et une leucoencéphalopathie a été confirmée. Une revue de la littérature portant sur 95 cas de mutations FOXC1 a montré que 6,3 % (6/95) présentaient des symptômes neuropsychiatriques, dont 83,3 % (5/6) avaient une mutation du domaine forkhead 2).
Cette observation suggère que le domaine fonctionnel de la mutation FOXC1 pourrait être impliqué dans l’apparition de symptômes neuropsychiatriques, soulignant l’importance d’un suivi à long terme du point de vue de la santé mentale.
Il a été suggéré que les mutations FOXC1 pourraient induire une CSVD et augmenter le risque d’accident vasculaire cérébral 4). La prévention et l’intervention précoce des maladies neurovasculaires chez les patients atteints d’ARS sont considérées comme des sujets de recherche futurs.
La compréhension de la pathologie de la « sclérisation » cornéenne due aux mutations FOXC1 progresse 1). On espère que la compréhension des mécanismes moléculaires de l’opacité cornéenne conduira au développement de nouveaux traitements utilisant la thérapie génique, les médicaments antifibrotiques et les biomatériaux 1).