Le syndrome de Sturge-Weber (SWS) est une neurocutanéose congénitale (phacomatose) également appelée angiomatose encéphalotrigéminée. Il se caractérise par la triade : angiome plan facial dans le territoire du trijumeau, angiome leptoméningé ipsilatéral et angiome oculaire. En 1879, Sturge a rapporté un cas d’hémiplégie et d’épilepsie associées à un angiome facial et un buphtalmie, et en 1929, Weber a établi le syndrome.
La cause est une mutation somatique en mosaïque du gène GNAQ (mutation post-zygotique), qui n’est pas héréditaire et survient principalement de manière sporadique4)8). On pense qu’elle est due à une anomalie du développement vasculaire causée par un trouble du système nerveux sympathique au cours de la période fœtale. La fréquence est rare, de 1 naissance sur 50 000, sans prédominance raciale ou de sexe5)7).
Le glaucome est la complication oculaire la plus importante du SWS, avec le taux d’incidence le plus élevé parmi les phacomatoses2). Lorsque l’hémangiome s’étend à la paupière, le glaucome survient avec une fréquence élevée de 30 à 70 %. Un hémangiome choroïdien est associé chez environ 40 % des patients.
Selon le moment de l’apparition, le glaucome est divisé en type précoce et type tardif. Environ 60 % sont de type précoce, survenant de la naissance à 4 ans, principalement dû à une anomalie du développement de l’angle. Les 40 % restants sont de type tardif, survenant après la petite enfance, impliquant une augmentation de la pression veineuse épisclérale et un hémangiome choroïdien. Il survient souvent avant l’âge de 10 ans.
QQu'est-ce que le syndrome de Sturge-Weber ?
A
Il s’agit d’une syndrome neurocutané congénital caractérisé par une triade : angiome plan facial dans le territoire du trijumeau, angiome leptoméningé homolatéral et angiome oculaire. La cause est une mutation somatique mosaïque du gène GNAQ, non héréditaire et le plus souvent sporadique4)8). Les principales complications sont des symptômes neurologiques (épilepsie chez 75 à 90 % des patients avant l’âge de 3 ans, déficience intellectuelle, hémiplégie) et un glaucome (30 à 70 %). Il existe également des types II (glaucome uniquement) et III (angiome méningé uniquement) sans la triade complète. Voir la section « Causes et facteurs de risque » pour plus de détails.
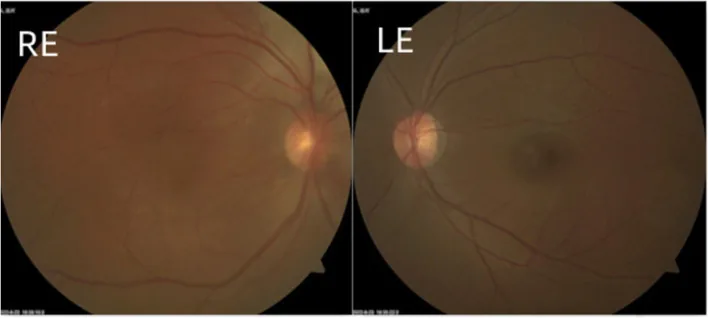
Zhang X, et al. Isolated diffuse choroidal hemangioma without systemic symptoms: a case report. BMC Ophthalmol. 2023. Figure 2. PMCID: PMC10324158. License: CC BY.
L’œil droit présente une couleur rouge diffuse plus intense que l’œil controlatéral, avec des lésions blanc-jaunâtres dispersées dans le pôle postérieur. Cela correspond à l’hémangiome choroïdien traité dans la section « 2. Principaux symptômes et signes cliniques ».
Dans la forme à début précoce, les premiers symptômes sont le larmoiement, la photophobie et le blépharospasme. Une augmentation du diamètre cornéen et une opacité cornéenne (buphthalmos) entraînent une déficience visuelle. Des stries de Haab (ruptures de la membrane de Descemet) peuvent être observées.
La forme à début tardif ressemble cliniquement au glaucome primitif à angle ouvert, avec peu de symptômes subjectifs au début. Avec la progression, un rétrécissement du champ visuel et une baisse de l’acuité visuelle apparaissent.
Signes cliniques (observés par le médecin lors de l’examen)
Glaucome : la complication oculaire la plus importante, survenant avec une fréquence élevée de 30 à 70 % lorsque l’hémangiome s’étend aux paupières1)
Hémangiome choroïdien : présent chez environ 40 % des patients. Le fond d’œil présente une rougeur diffuse (aspect « ketchup de tomate »). Peut entraîner un décollement de rétine exsudatif.
Angiographie à la fluorescéine : au début, on observe un motif de gros vaisseaux choroïdiens, et tardivement, une hyperfluorescence de toute la zone tumorale.
Dilatation et tortuosité des vaisseaux épiscléraux et conjonctivaux : observées comme signe d’augmentation de la pression veineuse épisclérale.
Signes à l’angle (forme à début précoce) : dysgénésie de l’angle avec insertion haute de la racine de l’iris.
Signes à l’angle (forme à début tardif) : anomalies angulaires minimes, mais du sang est souvent observé dans l’angle, suggérant une augmentation de la pression veineuse épisclérale.
Angiome plan (tache de vin) : Présent dès la naissance dans les territoires des première (V1) et deuxième (V2) branches du nerf trijumeau. Habituellement unilatéral mais peut être bilatéral7). La couleur s’assombrit et l’épaisseur augmente avec l’âge.
Crises d’épilepsie : Observées chez environ 80 % des patients. 75 à 90 % débutent avant l’âge de 3 ans. Des crises surviennent chez 95 % des patients présentant une atteinte leptoméningée bilatérale.
Symptômes neurologiques : Retard du développement psychomoteur (environ 50 %), hémiparésie controlatérale, hémianopsie latérale homonyme. L’angiome leptoméningé entraîne une atrophie et une calcification progressives du cortex cérébral.
Cas à début adulte : Un cas de première crise d’épilepsie à 55 ans a été rapporté5). Les cas atypiques sans angiome plan facial peuvent être diagnostiqués tardivement.
Le SWS est causé par une mutation somatique mosaïque du gène GNAQ (chromosome 9q21.2, c.548G→A, p.Arg183Gln)8). Cette mutation entraîne une activation constitutive de la voie de signalisation Gαq, conduisant à une prolifération incontrôlée des cellules endothéliales et à des malformations vasculaires4). Il s’agit d’une mutation somatique mosaïque, non germinale, donc non héréditaire. Le diagnostic moléculaire nécessite une biopsie du tissu atteint (généralement cutané)4).
La détection de la mutation GNAQ dépend de l’échantillon de tissu atteint et de la méthode d’analyse ; le diagnostic moléculaire doit donc être interprété en conjonction avec le tableau clinique.
Territoire V1 uniquement : Risque de glaucome 6,7 %
Territoire V2 uniquement : Risque de glaucome quasi nul
Territoires V1 + V2 : Risque de glaucome nettement augmenté à 31,8 %
Territoires V1 + V2 + V3 : Risque de symptômes neurologiques multiplié par 4
Différences selon l'âge d'apparition et l'étiologie
Forme précoce (environ 60 %) : Principalement due à une anomalie de développement de l’angle iridocornéen. Se manifeste par un œil de bœuf et une augmentation du diamètre cornéen1)
Forme tardive (environ 40 %) : Principalement due à une augmentation de la pression veineuse épisclérale et à l’implication d’un hémangiome choroïdien1)
Atteinte palpébrale : Lorsque l’hémangiome s’étend à la paupière, le risque de glaucome augmente considérablement
PWS bilatéral : Plus susceptible d’être associé à un SWS par rapport au PWS unilatéral7)
Le diagnostic du glaucome associé au SWS nécessite une mesure précise de la pression intraoculaire ainsi qu’un examen du segment antérieur, de l’angle iridocornéen et du fond d’œil. Chez l’enfant, un examen sous anesthésie générale est souvent indispensable.
Mesure de la pression intraoculaire : Chez l’enfant, le tonomètre de Goldmann est souvent difficile à utiliser ; le tonomètre à rebond (iCare, etc.) est utile. Il faut être prudent dans l’interprétation des mesures en cas d’œdème cornéen ou d’amincissement cornéen.
Examen du segment antérieur : Vérifier l’augmentation du diamètre cornéen (environ 10,5 mm chez le nouveau-né normal), l’opacité cornéenne et la présence de stries de Haab (fissures de la membrane de Descemet).
Examen de l’angle iridocornéen : Indispensable pour le diagnostic du type de glaucome et le choix du traitement. Dans la forme précoce, on observe une dysgénésie de l’angle (insertion anormalement haute de la racine de l’iris). Dans la forme tardive, du sang peut être observé dans l’angle, suggérant une augmentation de la pression veineuse épisclérale.
Examen du fond d’œil : Évaluer l’excavation de la papille optique (élargissement de l’excavation). En présence d’un hémangiome choroïdien, le fond d’œil peut présenter une teinte rouge diffuse.
L’angiographie à la fluorescéine est utile. On observe un motif vasculaire choroïdien large précocement, et une hyperfluorescence de toute la partie tumorale tardivement. L’hémangiome choroïdien diffus peut être difficile à identifier lors d’un examen du fond d’œil standard.
La tomodensitométrie crânienne détecte les calcifications intracorticales cérébrales. Même chez les nouveau-nés sans calcifications, l’IRM avec gadolinium peut détecter l’angiome leptoméningé. L’évaluation du débit sanguin cérébral par SPECT est également utilisée en complément.
Le SWS est une phacomatose et une maladie représentative provoquant un hémangiome choroïdien. Le diagnostic différentiel avec d’autres phacomatoses est important.
De plus, le syndrome de Klippel-Trenaunay-Weber présente des hémangiomes cutanés similaires au SWS, mais s’en distingue par la présence de malformations veineuses des membres et d’hypertrophie des tissus mous osseux.
QComment diagnostique-t-on le glaucome associé au SWS ?
A
Chez les patients présentant un angiome plan facial, un examen ophtalmologique régulier comprenant une mesure de la pression intraoculaire est essentiel. Chez l’enfant, on réalise une mesure de la pression intraoculaire au tonomètre à rebond, un examen du segment antérieur (diamètre cornéen, opacité cornéenne, présence de stries de Haab), une gonioscopie (évaluation des anomalies de l’angle), et un examen du fond d’œil (évaluation de l’excavation papillaire, détection d’hémangiome choroïdien). Une anesthésie générale est souvent nécessaire chez l’enfant. Dans les formes tardives, la présence de sang dans l’angle ou la dilatation des veines épisclérales est utile pour le diagnostic différentiel. L’angiographie à la fluorescéine est indispensable pour évaluer l’hémangiome choroïdien, et un scanner cérébral ou une IRM avec contraste sont réalisés pour l’évaluation systémique.
Le glaucome congénital ou débutant dans la petite enfance nécessite un traitement chirurgical 1). La trabéculotomie ou la goniotomie sont les interventions de première intention 1). Cependant, le taux de succès est inférieur à celui du glaucome congénital primitif, et des interventions supplémentaires sont souvent nécessaires.
La trabéculectomie comporte un risque d’hémorragie à partir de l’hémangiome, pouvant entraîner un hématome suprachoroïdien sévère ou une hémorragie expulsive. En général, la réponse à la chirurgie du glaucome est médiocre, et une trabéculectomie ou une chirurgie de drainage par tube sont souvent nécessaires.
Traitement des formes à début tardif (après la petite enfance)
Chez les patients plus âgés, la pression veineuse épisclérale étant élevée, le traitement médicamenteux est de première intention 1). Les inhibiteurs de la production d’humeur aqueuse (bêta-bloquants, inhibiteurs de l’anhydrase carbonique) sont considérés comme les plus efficaces. L’effet hypotenseur des prostaglandines est rapporté comme inconstant.
Traitement chirurgical (en cas d’échec médicamenteux)
Lorsque le traitement médicamenteux ou la reconstruction de la voie d’écoulement est inefficace, une trabéculectomie ou une chirurgie de shunt tubulaire peut être envisagée 1).
Chirurgie filtrante et shunt tubulaire
Trabéculectomie : L’utilisation concomitante d’antimétabolites (mitomycine C) peut améliorer les résultats. Cependant, dans les yeux atteints de SWS, le risque d’épanchement choroïdien et d’hémorragie expulsive est très élevé 1).
Dispositif Ahmed : Des études rapportent un taux de succès cumulé de 79 % à 24 mois et de 30 % à 60 mois.
Dispositif Baerveldt en deux étapes : Une étude rapporte que tous les cas ont atteint une pression intraoculaire inférieure à 21 mmHg avec un suivi moyen de 35 mois.
Méta-analyse des GDD pédiatriques : Analyse de 1 221 yeux montrant un taux de succès de 87 % (IC 95 % : 83-91 %) à 12 mois et de 77 % (IC 95 % : 71-83 %) à 24 mois 9).
Gestion des complications per- et postopératoires
Hémorragie et épanchement choroïdiens : En présence d’un hémangiome choroïdien, une baisse brutale de la pression intraoculaire augmente le risque de décollement et d’hémorragie choroïdiens 1).
Mesures préventives : Baisse préopératoire de la pression intraoculaire par des agents hyperosmotiques, réalisation d’une sclérotomie postérieure, mise en place préalable de sutures du volet scléral et sutures supplémentaires solides.
Choix du dispositif : Utilisation d’un GDD à valve (Ahmed) ou en deux étapes (Baerveldt) pour réduire le risque d’hypotonie.
Cyclophotocoagulation : Envisager une cyclophotocoagulation (CPC) dans les cas réfractaires. Une étude rapporte que 10 yeux sur 16 (62,5 %) ont maintenu une pression intraoculaire de 6 à 22 mmHg sans complications (suivi moyen de 8,87 ans).
Dans la méthode en deux étapes, une capsule se forme autour de la plaque plusieurs semaines avant l’insertion du tube dans la chambre antérieure. Cela évite une baisse excessive de la pression intraoculaire immédiatement après l’opération et minimise le risque d’épanchement et d’hémorragie choroïdiens. Cette technique est particulièrement utile dans les cas de SWS compliqués d’hémangiome choroïdien.
Il a été rapporté que le nétarsudil, même ajouté comme traitement de 4e ou 5e intention, abaisse efficacement la pression intraoculaire dans le glaucome associé au SWS. Il agit en favorisant l’écoulement de l’humeur aqueuse par le trabéculum.
L’hémangiome choroïdien diffus associé au SWS peut nécessiter une prise en charge parallèle au traitement du glaucome.
Asymptomatique : Surveillance
Baisse de la fonction visuelle due à un décollement séreux de la rétine : Photocoagulation rétinienne, thermothérapie transpupillaire (TTT) ou thérapie photodynamique (PDT) peuvent être envisagées. Cependant, la PDT n’est pas remboursée par l’assurance maladie.
Bien que plus réfractaire que le glaucome congénital, un bon contrôle précoce de la pression intraoculaire peut permettre de préserver la vision. Cependant, si l’hémangiome choroïdien s’agrandit et provoque un décollement de rétine exsudatif, la cryocoagulation peut ne pas être suffisamment efficace et entraîner une déficience visuelle sévère.
En raison de l’atteinte de la cornée, du cristallin, de la rétine et du nerf optique, l’obtention d’une bonne acuité visuelle est souvent difficile. L’espérance de vie des patients atteints de SWS est réduite par rapport à la population générale, et en cas de lésion leptoméningée bilatérale, les symptômes neurologiques sont plus graves et le pronostic est plus sombre.
QQuels sont les points particulièrement importants dans le traitement du glaucome du SWS ?
A
Le point le plus important concerne les complications chirurgicales liées à l’hémangiome choroïdien. Dans le SWS, un hémangiome choroïdien est présent dans environ 40 % des cas, et une baisse brutale de la pression intraoculaire lors de la chirurgie du glaucome comporte un risque d’épanchement choroïdien, d’hémorragie et de décollement de rétine1). Les mesures préventives recommandées comprennent l’administration préopératoire d’agents hyperosmotiques, la réalisation d’une sclérotomie postérieure, une suture solide du volet scléral et l’utilisation d’un dispositif de drainage à valve ou en deux étapes. Il faut également noter que les agonistes des récepteurs alpha-2 (brimonidine) sont contre-indiqués chez les enfants de moins de 2 ans1). Pour plus de détails, voir la section « Traitement standard ».
La cause fondamentale du SWS est une mutation somatique en mosaïque du gène GNAQ (c.548G→A, p.Arg183Gln)8). Cette mutation active de manière constitutive la voie de signalisation Gαq, entraînant une prolifération incontrôlée des cellules endothéliales et des malformations vasculaires4). La mutation survenant dans les cellules somatiques tôt après la fécondation, la distribution des cellules mutées détermine la diversité des phénotypes cliniques (types I à III).
Mécanisme de l’élévation de la pression intraoculaire
Selon les directives cliniques pour le glaucome (5e édition), les mécanismes suivants sont énumérés pour l’élévation de la pression intraoculaire dans le SWS1).
Dysgénésie de l’angle primaire : anomalie congénitale du développement de la voie d’écoulement de l’humeur aqueuse, principale cause des formes à début précoce
Atrophie du canal de Schlemm : anomalie structurelle de la voie d’écoulement contribuant à l’élévation de la pression intraoculaire1)
Augmentation de la pression veineuse épisclérale : les hémangiomes de la surface oculaire et de l’orbite perturbent le retour veineux, augmentant la résistance à l’écoulement de l’humeur aqueuse. C’est la cause principale des formes à début tardif1)
Formation de synéchies antérieures périphériques (PAS) : les néovaisseaux de l’iris et de l’angle peuvent entraîner un glaucome secondaire par fermeture de l’angle2)3)
Augmentation de la perméabilité des parois vasculaires amincies liée à l’hémangiome choroïdien : l’exsudation de la choroïde contribue à l’élévation de la pression intraoculaire1)
Résultats histologiques des formes à début précoce (dysgénésie de l’angle)
Les examens histologiques d’yeux énucléés ont révélé les résultats suivants, similaires à ceux observés dans le glaucome congénital primaire.
Trabéculum uvéal large
Muscle ciliaire attaché directement au trabéculum
Éperon scléral sous-développé
Racine de l’iris attachée antérieurement
Lorsque la maladie se déclare dans la petite enfance, une anomalie de développement de l’angle iridocornéen est considérée comme le facteur le plus important.
Mécanisme du type à apparition tardive (augmentation de la pression veineuse épisclérale)
Lorsque la maladie se déclare à la fin de l’adolescence ou après la vingtaine, l’augmentation de la pression veineuse épisclérale due à un angiome est la cause principale 1). À la gonioscopie, l’anomalie de l’angle est minime, mais du sang est souvent observé dans l’angle, ce qui est corrélé à l’augmentation de la pression veineuse épisclérale. Dans cette condition, les inhibiteurs de la production d’humeur aqueuse sont considérés comme les plus efficaces, et le traitement médicamenteux est la première option.
Vieillissement précoce du trabéculum : proposé comme mécanisme provoquant un glaucome chronique à angle ouvert chez le jeune adulte
Pathologie de l’hémangiome choroïdien : une prolifération anormale des vaisseaux dans la choroïde provoque des lésions surélevées rouges à rouge-orange au fond d’œil. Ceux associés au SWS sont souvent diffus et mal délimités, présentant un tableau clinique différent de celui de l’hémangiome choroïdien solitaire.
Pathologie cérébrale : l’hémangiome leptoméningé provoque une stase veineuse ; initialement, la perfusion sanguine augmente de manière compensatoire, mais ensuite le débit sanguin cérébral et le métabolisme du glucose diminuent, entraînant une dégénérescence et une atrophie neuronales progressives.
QPourquoi les traitements diffèrent-ils entre le type à apparition précoce et le type à apparition tardive ?
A
Cela est dû au fait que les mécanismes de développement du glaucome sont fondamentalement différents. Dans le type à apparition précoce, une anomalie congénitale de l’angle iridocornéen est la cause principale ; il existe un problème au niveau du trabéculum ou de la structure de l’angle elle-même, donc la chirurgie de l’angle (trabéculotomie, goniotomie) qui ouvre physiquement la voie d’écoulement est efficace 1). En revanche, dans le type à apparition tardive, l’augmentation de la pression veineuse épisclérale est la cause principale, et la structure de l’angle elle-même est relativement normale. Lorsque la pression veineuse épisclérale est élevée, les inhibiteurs de la production d’humeur aqueuse sont les plus efficaces, et le traitement médicamenteux est la première option 1). Si le traitement médicamenteux est inefficace, on envisage une chirurgie filtrante ou une chirurgie de tube de dérivation qui peut contourner le système veineux épiscléral. Voir la section « Traitement standard » pour plus de détails.
Dans la gestion du glaucome associé au SWS, des progrès sont attendus dans les domaines suivants.
Diagnostic moléculaire de la mutation somatique en mosaïque de GNAQ : la mutation GNAQ identifiée par Shirley et al. en 2013 a clarifié la base moléculaire du SWS et de l’hémangiome plan 8). À l’avenir, un diagnostic précoce basé sur des tests génétiques et un traitement personnalisé sont attendus.
Dispositif de drainage du glaucome en deux étapes : L’utilité du dispositif Baerveldt en deux étapes a été rapportée comme une méthode pour réduire le risque de complications choroïdiennes associées à la chirurgie filtrante conventionnelle.
Nétarsudil (inhibiteur de la Rho kinase) : En tant que médicament favorisant l’écoulement de l’humeur aqueuse du trabéculum, son efficacité a été rapportée dans le glaucome associé au SWS.
Importance de la collaboration pluridisciplinaire : La collaboration entre pédiatrie, neurologie, ophtalmologie, dermatologie et psychiatrie est indispensable pour la prise en charge à long terme du SWS6). Il a également été rapporté que les complications psychiatriques influencent les résultats chirurgicaux, et l’importance des soins complets incluant un soutien psychologique est reconnue.
Traitement au laser à colorant pulsé pour les angiomes plans : Un début précoce est recommandé, mais la disparition complète de la décoloration cutanée est rare. Le traitement est considéré comme plus favorable pour les taches de la partie centrale du front que pour celles de la partie centrale du visage.
European Glaucoma Society. European Glaucoma Society Terminology and Guidelines for Glaucoma, 5th Edition. Br J Ophthalmol. 2021 Jun;105(Suppl 1):1-169. doi:10.1136/bjophthalmol-2021-egsguidelines. PMID:34675001.
Pazos M, Traverso CE, Viswanathan A; European Glaucoma Society. European Glaucoma Society - Terminology and guidelines for glaucoma, 6th Edition. Br J Ophthalmol. 2025;109(Suppl 1):1-212. doi:10.1136/bjophthalmol-2025-egsguidelines. PMID:41026937.
Yadav PS, Adhikari P, Mehta B, Khadka S, Bhurtel MR, Dahal A, et al. Unmasking Sturge-Weber syndrome in adulthood: a case with extrafacial port-wine stain and delayed neurological symptoms. Annals of medicine and surgery (2012). 2024;86(6):3679-3682. doi:10.1097/MS9.0000000000002049. PMID:38846877; PMCID:PMC11152852.
Ainuz BY, Wolfe EM, Wolfe SA. Surgical Management of Facial Port-Wine Stain in Sturge Weber Syndrome. Cureus. 2021;13(1):e12637. doi:10.7759/cureus.12637. PMID:33585124; PMCID:PMC7872872.
Pathak BD, Sharma S, Adhikari A, Simkhada N, Ghimire B, Aryal N. Sturge-Weber Syndrome with Bilateral Port-Wine Stain. Case reports in pediatrics. 2022;2022:2191465. doi:10.1155/2022/2191465. PMID:35464665; PMCID:PMC9033375.
Shirley MD, Tang H, Gallione CJ, Baugher JD, Frelin LP, Cohen B, et al. Sturge-Weber syndrome and port-wine stains caused by somatic mutation in GNAQ. The New England journal of medicine. 2013;368(21):1971-9. doi:10.1056/NEJMoa1213507. PMID:23656586; PMCID:PMC3749068.
Stallworth JY, O’Brien KS, Han Y, Oatts JT. Efficacy of Ahmed and Baerveldt glaucoma drainage device implantation in the pediatric population: A systematic review and meta-analysis. Surv Ophthalmol. 2023;68(4):616-629.
Copiez le texte de l'article et collez-le dans l'assistant IA de votre choix.
Article copié dans le presse-papiers
Ouvrez un assistant IA ci-dessous et collez le texte copié dans la conversation.