Verteilung des Feuermals und Risiko
Nur V1-Areal : Glaukomrisiko 6,7 %
Nur V2-Areal : Glaukomrisiko nahezu null
V1 + V2-Areal : Glaukomrisiko deutlich erhöht auf 31,8 %
V1 + V2 + V3-Areal : Risiko für neurologische Symptome 4-fach erhöht

Das Sturge-Weber-Syndrom (SWS) ist ein angeborenes neurokutanes Syndrom (Phakomatose), das auch als enzephalotrigeminale Angiomatose bezeichnet wird. Es ist gekennzeichnet durch die Trias: Gesichts-Feuermal (Naevus flammeus) im Bereich des Trigeminusnervs, ipsilaterales leptomeningeales Angiom und okuläres Angiom. 1879 berichtete Sturge über einen Fall von Hemiplegie und Epilepsie in Verbindung mit einem Gesichtsangiom und Buphthalmus, und 1929 etablierte Weber das Syndrom.
Die Ursache ist eine somatische Mosaikmutation des GNAQ-Gens (postzygotische Mutation), die nicht vererbt wird und meist sporadisch auftritt4)8). Es wird angenommen, dass sie auf eine abnorme Gefäßentwicklung aufgrund einer Störung des sympathischen Nervensystems während der Embryonalzeit zurückzuführen ist. Die Häufigkeit ist selten, 1 von 50.000 Geburten, ohne Bevorzugung von Rasse oder Geschlecht5)7).
Das SWS wird anhand der klinischen Merkmale in die folgenden drei Typen eingeteilt.
| Klassifikation (Roach) | Merkmale |
|---|---|
| Typ I (klassisch) | PWS + neurologische Symptome + Glaukom |
| Typ II | PWS + Glaukom (ohne neurologische Läsion) |
| Typ III (sehr selten) | Nur meningeales Angiom |
Das Glaukom ist die wichtigste Augenkomplikation des SWS und weist die höchste Glaukom-Inzidenzrate unter den Phakomatosen auf2). Wenn das Hämangiom das Augenlid betrifft, tritt ein Glaukom mit einer hohen Häufigkeit von 30–70 % auf. Ein Aderhaut-Hämangiom tritt bei etwa 40 % der Patienten auf.
Je nach Zeitpunkt des Auftretens wird das Glaukom in einen frühen und einen späten Typ unterteilt. Etwa 60 % sind vom frühen Typ, der von der Geburt bis zum Alter von 4 Jahren auftritt, hauptsächlich aufgrund einer Entwicklungsanomalie des Kammerwinkels. Die restlichen etwa 40 % sind vom späten Typ, der nach dem Säuglingsalter auftritt, wobei ein erhöhter episkleraler Venendruck und ein Aderhaut-Hämangiom eine Rolle spielen. Es tritt häufig vor dem 10. Lebensjahr auf.
Es handelt sich um ein angeborenes neurokutanes Syndrom mit der Trias: Gesichtsangiom im Trigeminusbereich, ipsilaterales leptomeningeales Angiom und okuläres Angiom. Ursache ist eine somatische Mosaikmutation im GNAQ-Gen, nicht erblich und meist sporadisch4)8). Hauptkomplikationen sind neurologische Symptome (Epilepsie bei 75–90 % vor dem 3. Lebensjahr, geistige Behinderung, Hemiparese) und Glaukom (30–70 %). Es gibt auch Typ II (nur Glaukom) und Typ III (nur meningeales Angiom) ohne vollständige Trias. Siehe Abschnitt „Ursachen und Risikofaktoren“ für Details.
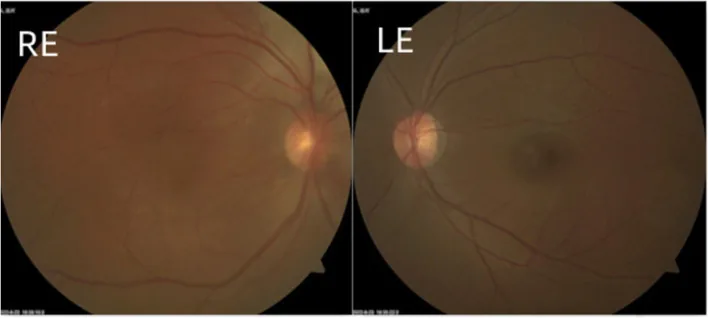
Bei der früh beginnenden Form sind Tränenfluss, Lichtscheu und Lidkrampf die ersten Symptome. Eine Vergrößerung des Hornhautdurchmessers und Hornhauttrübung (Buphthalmus) führen zu Sehstörungen. Es können Haab-Linien (Risse in der Descemet-Membran) auftreten.
Die spät beginnende Form ähnelt klinisch dem primären Offenwinkelglaukom mit anfangs geringen subjektiven Symptomen. Im Verlauf treten Gesichtsfeldeinschränkungen und Sehverschlechterung auf.
SWS wird durch eine somatische Mosaikmutation im GNAQ-Gen (Chromosom 9q21.2, c.548G→A, p.Arg183Gln) verursacht8). Diese Mutation führt zu einer konstitutiven Aktivierung des Gαq-Signalwegs, was eine unkontrollierte Proliferation von Endothelzellen und vaskuläre Fehlbildungen zur Folge hat4). Da es sich um eine somatische Mosaikmutation und nicht um eine Keimbahnmutation handelt, ist sie nicht vererbbar. Für die molekulare Diagnostik ist eine Biopsie des betroffenen Gewebes (meist Haut) erforderlich4).
Der Nachweis der GNAQ-Mutation hängt von der Gewebeprobe und der Analysemethode ab; daher sollte die molekulare Diagnostik in Zusammenschau mit dem klinischen Bild interpretiert werden.
Verteilung des Feuermals und Risiko
Nur V1-Areal : Glaukomrisiko 6,7 %
Nur V2-Areal : Glaukomrisiko nahezu null
V1 + V2-Areal : Glaukomrisiko deutlich erhöht auf 31,8 %
V1 + V2 + V3-Areal : Risiko für neurologische Symptome 4-fach erhöht
Unterschiede im Erkrankungsalter und in der Ätiologie
Frühmanifestation (ca. 60 %) : Hauptsächlich durch eine Entwicklungsstörung des Kammerwinkels. Zeigt sich mit Buphthalmus und vergrößertem Hornhautdurchmesser1)
Spätmanifestation (ca. 40 %) : Hauptsächlich durch erhöhten episkleralen Venendruck und Beteiligung eines Aderhautangioms1)
Lidbeteiligung : Wenn das Angiom auf das Lid übergreift, steigt die Glaukom-Inzidenz deutlich an
Bilaterales PWS : Im Vergleich zu unilateralem PWS besteht eine höhere Wahrscheinlichkeit für ein SWS7)
Die Diagnose eines mit SWS assoziierten Glaukoms erfordert eine präzise Augeninnendruckmessung sowie eine Untersuchung des vorderen Augenabschnitts, des Kammerwinkels und des Augenhintergrunds. Bei Kindern ist eine Untersuchung in Vollnarkose oft unerlässlich.
Die Fluoreszenzangiographie ist nützlich. Frühzeitig zeigt sich ein großes choroidales Gefäßmuster, und spät zeigt der gesamte Tumorbereich eine Hyperfluoreszenz. Das diffuse Aderhautangiom kann bei der normalen Fundusuntersuchung schwer zu identifizieren sein.
Die CT des Kopfes erkennt Verkalkungen in der Hirnrinde. Auch bei Neugeborenen, bei denen noch keine Verkalkungen aufgetreten sind, kann ein leptomeningeales Angiom mittels Gadolinium-verstärkter MRT nachgewiesen werden. Die Beurteilung der zerebralen Durchblutung mittels SPECT wird ergänzend eingesetzt.
Das SWS ist eine Phakomatose und eine repräsentative Erkrankung, die ein Aderhautangiom verursacht. Die Abgrenzung zu anderen Phakomatosen ist wichtig.
| Erkrankung | Charakteristische Augenveränderungen | Lokalisation des Angioms/Tumors |
|---|---|---|
| SWS | Aderhautangiom, Glaukom | Gesicht + Leptomeninx |
| Von-Hippel-Lindau-Erkrankung | Retinahämangiom (temporale Peripherie) | Retina, Kleinhirn, Niere |
| Neurofibromatose Typ 1 | Irische Lisch-Knötchen | Kutane Neurofibrome |
| Tuberöse Sklerose | Retinales Hamartom | Gehirn, Haut, Nieren, Herz |
Das Klippel-Trenaunay-Weber-Syndrom zeigt ebenfalls kutane Hämangiome ähnlich dem SWS, unterscheidet sich jedoch durch das Vorliegen von venösen Malformationen der Extremitäten und Hypertrophie der Knochen- und Weichteile.
Bei Patienten mit fazialem Feuermal sind regelmäßige augenärztliche Untersuchungen einschließlich Augeninnendruckmessung unerlässlich. Bei Kindern werden die Augeninnendruckmessung mit einem Rückpralltonometer, die Untersuchung des vorderen Augenabschnitts (Hornhautdurchmesser, Hornhauttrübung, Vorhandensein von Haab-Linien), die Gonioskopie (Beurteilung von Kammerwinkelanomalien) und die Fundusuntersuchung (Beurteilung der Papillenexkavation, Nachweis von Aderhauthämangiomen) durchgeführt. Bei Kindern ist häufig eine Untersuchung in Vollnarkose erforderlich. Bei spätmanifesten Formen sind Blut im Kammerwinkel oder erweiterte episklerale Venen für die Differentialdiagnose hilfreich. Zur Beurteilung von Aderhauthämangiomen ist eine Fluoreszenzangiographie unverzichtbar, und zur systemischen Beurteilung werden ein CT oder MRT mit Kontrastmittel des Kopfes durchgeführt.
Die Behandlung des Glaukoms bei SWS erfordert je nach Manifestationsalter und Pathomechanismus unterschiedliche Strategien.
Das angeborene oder im Säuglingsalter auftretende Glaukom erfordert eine chirurgische Behandlung 1). Die Trabekulotomie oder Goniotomie sind die Erstlinienverfahren 1). Die Erfolgsrate ist jedoch geringer als beim primären kongenitalen Glaukom, und häufig sind zusätzliche Eingriffe erforderlich.
Eine Trabekulektomie birgt das Risiko von Blutungen aus dem Hämangiom, die zu einem schweren suprachoroidalen Hämatom oder einer expulsiven Blutung führen können. Im Allgemeinen spricht das Glaukom schlecht auf Operationen an, und häufig sind eine Trabekulektomie oder eine Tube-Shunt-Operation erforderlich.
Bei älteren Kindern ist der episklerale Venendruck erhöht, daher ist die medikamentöse Therapie die erste Wahl 1). Medikamente, die die Kammerwasserproduktion hemmen (Betablocker, Carboanhydrasehemmer), gelten als am wirksamsten. Die drucksenkende Wirkung von Prostaglandinanaloga wird als inkonsistent beschrieben.
Wenn die medikamentöse Behandlung oder die Wiederherstellung des Abflusswegs nicht erfolgreich ist, kann eine Trabekulektomie oder eine Tubus-Shunt-Operation in Betracht gezogen werden 1).
Fistulierende Operation und Tubus-Shunt
Trabekulektomie : Die gleichzeitige Anwendung von Antimetaboliten (Mitomycin C) kann die Ergebnisse verbessern. Bei SWS-Augen ist das Risiko eines Aderhautergusses und einer expulsiven Blutung jedoch sehr hoch 1).
Ahmed-Implantat : Berichten zufolge beträgt die kumulative Erfolgsrate nach 24 Monaten 79 % und nach 60 Monaten 30 %.
Zweistufiges Baerveldt-Implantat : Eine Studie berichtet, dass bei einer durchschnittlichen Nachbeobachtungszeit von 35 Monaten alle Fälle einen Augeninnendruck unter 21 mmHg erreichten.
Metaanalyse zu pädiatrischen GDD : Analyse von 1.221 Augen ergab eine Erfolgsrate von 87 % (95 %-KI: 83-91 %) nach 12 Monaten und 77 % (95 %-KI: 71-83 %) nach 24 Monaten 9).
Management intra- und postoperativer Komplikationen
Aderhautblutung und -erguss : Bei Vorliegen eines Aderhauthämangioms erhöht ein plötzlicher Abfall des Augeninnendrucks das Risiko einer Aderhautablösung und -blutung 1).
Präventive Maßnahmen : Präoperativer Augeninnendrucksenkung durch hyperosmotische Mittel, Durchführung einer hinteren Sklerotomie, Vorsetzen von Skleralappen-Nähten und feste zusätzliche Nähte.
Implantatauswahl : Verwendung eines ventilierten (Ahmed) oder zweistufigen (Baerveldt) GDD reduziert das Risiko einer Hypotonie.
Zyklophotokoagulation : Bei therapierefraktären Fällen ist eine Zyklophotokoagulation (CPC) in Betracht zu ziehen. Ein Bericht zeigt, dass 10 von 16 Augen (62,5 %) ohne Komplikationen einen Augeninnendruck von 6–22 mmHg aufrechterhielten (durchschnittliche Nachbeobachtungszeit 8,87 Jahre).
Bei der zweistufigen Methode wird einige Wochen vor dem Einsetzen des Tubus in die Vorderkammer eine Kapsel um die Platte gebildet. Dies verhindert einen übermäßigen Augeninnendruckabfall unmittelbar nach der Operation und minimiert das Risiko von Aderhauterguss und -blutung. Diese Technik ist besonders nützlich bei SWS-Fällen, die mit einem Aderhauthämangiom kompliziert sind.
Es wurde berichtet, dass Netarsudil, selbst wenn es als Medikament der 4. oder 5. Wahl hinzugefügt wird, den Augeninnendruck bei Glaukom im Zusammenhang mit SWS wirksam senkt. Es wirkt, indem es den Abfluss des Kammerwassers durch das Trabekelwerk fördert.
Das diffuse Aderhautangiom im Rahmen des SWS muss möglicherweise parallel zur Glaukombehandlung behandelt werden.
Obwohl es schwieriger zu behandeln ist als das angeborene Glaukom, kann bei frühzeitiger guter Augeninnendruckkontrolle das Sehvermögen erhalten werden. Wenn sich das Aderhautangiom jedoch vergrößert und zu einer exsudativen Netzhautablösung führt, kann selbst eine Kryokoagulation nicht ausreichend wirksam sein und zu einer schweren Sehbehinderung führen.
Aufgrund der Beteiligung von Hornhaut, Linse, Netzhaut und Sehnerv ist das Erreichen einer guten Sehschärfe oft schwierig. Die Lebenserwartung von SWS-Patienten ist im Vergleich zur Allgemeinbevölkerung verkürzt, und bei beidseitigen leptomeningealen Läsionen sind die neurologischen Symptome schwerwiegender und die Prognose schlechter.
Der wichtigste Punkt sind die operationsbedingten Komplikationen im Zusammenhang mit dem Aderhautangiom. Beim SWS liegt in etwa 40 % der Fälle ein Aderhautangiom vor, und ein plötzlicher Abfall des Augeninnendrucks während der Glaukomoperation birgt das Risiko eines Aderhautergusses, einer Blutung und einer Netzhautablösung1). Als vorbeugende Maßnahmen werden die präoperative Gabe von hyperosmotischen Mitteln, die Durchführung einer hinteren Sklerotomie, eine feste Naht des Skleradeckels und die Verwendung eines ventilierten oder zweistufigen Drainage-Implantats empfohlen. Beachten Sie auch, dass Alpha-2-Rezeptor-Agonisten (Brimonidin) bei Kindern unter 2 Jahren kontraindiziert sind1). Einzelheiten finden Sie im Abschnitt „Standardbehandlung“.
Die grundlegende Ursache des SWS ist eine somatische Mosaikmutation im GNAQ-Gen (c.548G→A, p.Arg183Gln)8). Diese Mutation aktiviert konstitutiv den Gαq-Signalweg, was zu unkontrollierter Proliferation von Endothelzellen und Gefäßfehlbildungen führt4). Da die Mutation in somatischen Zellen kurz nach der Befruchtung auftritt, bestimmt die Verteilung der mutierten Zellen die Vielfalt der klinischen Phänotypen (Typ I–III).
Gemäß der Leitlinie zur Glaukomdiagnostik (5. Auflage) werden für den Augeninnendruckanstieg bei SWS die folgenden fünf Mechanismen aufgeführt1).
Histologische Untersuchungen enukleierter Augen haben folgende Befunde ergeben, die denen des primären kongenitalen Glaukoms ähneln.
Wenn die Erkrankung im Säuglingsalter auftritt, wird eine Entwicklungsanomalie des Kammerwinkels als wichtigster Faktor angesehen.
Wenn die Erkrankung im späten Teenageralter oder nach dem 20. Lebensjahr auftritt, ist ein erhöhter episkleraler Venendruck aufgrund eines Angioms die Hauptursache 1). In der Gonioskopie ist die Winkelanomalie minimal, aber im Winkel wird häufig Blut beobachtet, was mit dem erhöhten episkleralen Venendruck korreliert. In diesem Zustand gelten Hemmstoffe der Kammerwasserproduktion als am wirksamsten, und die medikamentöse Therapie ist die erste Wahl.
Dies liegt daran, dass die Mechanismen der Glaukomentstehung grundlegend unterschiedlich sind. Beim früh einsetzenden Typ ist eine angeborene Entwicklungsanomalie des Kammerwinkels die Hauptursache; es liegt ein Problem mit dem Trabekelwerk oder der Winkelstruktur selbst vor, daher ist eine Winkelchirurgie (Trabekulotomie, Goniotomie), die den Abflussweg physisch öffnet, wirksam 1). Beim spät einsetzenden Typ hingegen ist ein erhöhter episkleraler Venendruck die Hauptursache, und die Winkelstruktur selbst ist relativ normal. Bei hohem episkleralen Venendruck sind Hemmstoffe der Kammerwasserproduktion am wirksamsten, und die medikamentöse Therapie ist die erste Wahl 1). Wenn die medikamentöse Therapie unwirksam ist, wird eine filtrierende Operation oder eine Tubus-Shunt-Operation in Betracht gezogen, die das episklerale Venensystem umgehen kann. Einzelheiten finden Sie im Abschnitt „Standardbehandlung“.
Im Management des mit SWS assoziierten Glaukoms werden Fortschritte in folgenden Bereichen erwartet.
日本緑内障学会緑内障診療ガイドライン改訂委員会. 緑内障診療ガイドライン(第5版). 日眼会誌. 2022;126(2):85-177.
European Glaucoma Society. European Glaucoma Society Terminology and Guidelines for Glaucoma, 5th Edition. Br J Ophthalmol. 2021 Jun;105(Suppl 1):1-169. doi:10.1136/bjophthalmol-2021-egsguidelines. PMID:34675001.
Pazos M, Traverso CE, Viswanathan A; European Glaucoma Society. European Glaucoma Society - Terminology and guidelines for glaucoma, 6th Edition. Br J Ophthalmol. 2025;109(Suppl 1):1-212. doi:10.1136/bjophthalmol-2025-egsguidelines. PMID:41026937.
Yeom S, Comi AM. Updates on Sturge-Weber Syndrome. Handb Clin Neurol. 2015;132:157-168. doi:10.1016/B978-0-444-62702-5.00011-1.
Yadav PS, Adhikari P, Mehta B, Khadka S, Bhurtel MR, Dahal A, et al. Unmasking Sturge-Weber syndrome in adulthood: a case with extrafacial port-wine stain and delayed neurological symptoms. Annals of medicine and surgery (2012). 2024;86(6):3679-3682. doi:10.1097/MS9.0000000000002049. PMID:38846877; PMCID:PMC11152852.
Ainuz BY, Wolfe EM, Wolfe SA. Surgical Management of Facial Port-Wine Stain in Sturge Weber Syndrome. Cureus. 2021;13(1):e12637. doi:10.7759/cureus.12637. PMID:33585124; PMCID:PMC7872872.
Pathak BD, Sharma S, Adhikari A, Simkhada N, Ghimire B, Aryal N. Sturge-Weber Syndrome with Bilateral Port-Wine Stain. Case reports in pediatrics. 2022;2022:2191465. doi:10.1155/2022/2191465. PMID:35464665; PMCID:PMC9033375.
Shirley MD, Tang H, Gallione CJ, Baugher JD, Frelin LP, Cohen B, et al. Sturge-Weber syndrome and port-wine stains caused by somatic mutation in GNAQ. The New England journal of medicine. 2013;368(21):1971-9. doi:10.1056/NEJMoa1213507. PMID:23656586; PMCID:PMC3749068.
Stallworth JY, O’Brien KS, Han Y, Oatts JT. Efficacy of Ahmed and Baerveldt glaucoma drainage device implantation in the pediatric population: A systematic review and meta-analysis. Surv Ophthalmol. 2023;68(4):616-629.