ARS тип 1
Причинный ген: PITX2 (4q25)
Основные аномалии: аномалии переднего отрезка глаза, аномалии зубов, избыток кожи вокруг пупка / пупочная грыжа, черепно-лицевые аномалии, сердечно-сосудистые аномалии

Синдром Аксельфельда-Ригера (ARS) — это группа врожденных заболеваний, сочетающих аномалии переднего сегмента глаза и системные нарушения. Основная причина — нарушение миграции и дифференцировки клеток нервного гребня. В конце эмбрионального периода нарушается нормальный процесс исчезновения недифференцированных эндотелиальных клеток, покрывающих переднюю камеру, из радужки и угла; их остатки приводят к образованию тяжей и высокому прикреплению радужки.
Историческая справка: В 1920 году Аксельфельд описал заднее эмбриональное кольцо (смещение вперед и утолщение линии Швальбе) и отростки радужки. В 1934–1935 годах Ригер добавил гипоплазию радужки, девиацию зрачка и поликорию. В настоящее время выделяют три стадии:
Все вместе это называется синдромом Аксельфельда-Ригера. В 50–60% случаев развивается глаукома, наследование аутосомно-доминантное, обычно двустороннее. Катаракта и эктопия хрусталика также часто сочетаются.
Эпидемиология: распространенность ранее оценивалась примерно как 1/200 000, но недавние сообщения предполагают 1/50 000–100 0002)4). Гендерных различий нет, диагноз часто ставится в младенческом или раннем детском возрасте.
Генетическая классификация:
ARS тип 1
Причинный ген: PITX2 (4q25)
Основные аномалии: аномалии переднего отрезка глаза, аномалии зубов, избыток кожи вокруг пупка / пупочная грыжа, черепно-лицевые аномалии, сердечно-сосудистые аномалии
ARS тип 2
Причинный ген: 13q14 (не подтвержден)
Основные аномалии: аномалии переднего отрезка глаза, глаукома. Системные аномалии встречаются реже, чем при типах 1 и 3.
ARS тип 3
Причинный ген: FOXC1 (6p25)
Основные аномалии: аномалии переднего отрезка глаза, глаукома, нейросенсорная тугоухость, дефект межпредсердной перегородки, аномалии почек, поражения белого вещества
Мутации FOXC1 и PITX2 составляют 40–70% случаев ARS5). Однако в 60% случаев ARS причинный ген не идентифицирован4), что указывает на большое генетическое разнообразие.
Анализ крупного регистра детской и ювенильной глаукомы показал частоту молекулярной диагностики 56,5%11). Мутации FOXC1 составили 20,3%, PITX2 – 17,4%, PAX6 – 10,1%, и немало случаев не могут быть объяснены известными генами11).
Они различаются по причинному гену. Тип 1 вызывается мутацией PITX2 (4q25) и сопровождается аномалиями зубов, пупка и лицевых костей. Тип 3 вызывается мутацией FOXC1 (6p25) и сопровождается тугоухостью, пороками сердца, аномалиями почек и неврологическими нарушениями. Тип 2 локализован на 13q14, но причинный ген не подтвержден; в основном проявляется аномалиями переднего отрезка глаза и глаукомой. Окончательный диагноз может быть поставлен с помощью генетического тестирования.
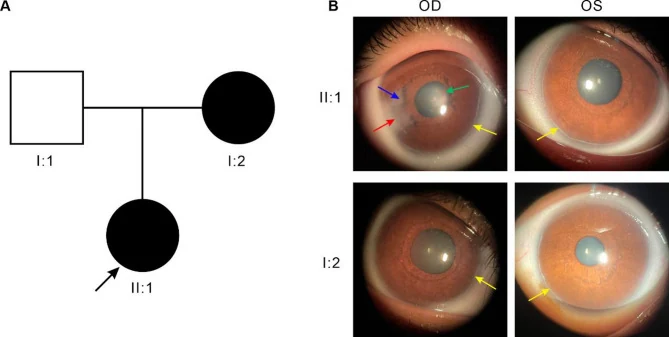
Основные глазные признаки приведены ниже.
| Глазной признак | Характеристики |
|---|---|
| Заднее эмбриональное кольцо | Смещение кпереди и утолщение линии Швальбе |
| Отростки радужки | Тонкие нитевидные до широких лентовидных |
| Смещение зрачка | Смещение в направлении, противоположном заднему эмбриональному кольцу |
| Псевдополикория | Перфоративный вид стромы радужки |
| Увеальный выворот | Выворот пигментного эпителия радужки |
Заднее эмбриональное кольцо представляет собой остаток недифференцированных клеток в области линии Швальбе, видимый в виде линии вдоль лимба, смещенной на 0,5–2,0 мм к центру. Часто оно не циркулярное, а ограничено участком. Если выступающая линия Швальбе срастается с радужкой, это называется аномалией Аксенфельда; если дополнительно присутствует атрофия стромы радужки — аномалией Ригера.
Роговица обычно прозрачна, эндотелий нормален, но физический контакт с остаточной тканью может вызвать вторичное помутнение роговицы. Помутнение часто ограничено периферией и обычно напрямую не влияет на зрение. Однако при мутациях FOXC1 помутнение роговицы и неоваскуляризация более выражены, степень аномалии роговицы больше, а частота глаукомы выше по сравнению с мутациями PITX21).
Угловые признаки включают высокое прикрепление радужки, тяжистые увеальные остатки и утолщение линии Швальбе (заднее эмбриональное кольцо). Сообщается также о случаях с микроофакией и подвывихом хрусталика7).
Глаукома встречается в 50–60% случаев. Повышение внутриглазного давления может возникнуть уже в младенчестве, но чаще всего оно проявляется в детском или молодом возрасте. Некоторые случаи диагностируются при прогрессирующем снижении зрения; важно не пропустить признаки переднего отрезка и глаукоматозные изменения6).
Li et al. (2021) сообщили о 7-летнем мальчике (ARS тип 3, мутация de novo FOXC1) с диаметром роговицы 14 мм, аксиальной длиной 27,16/26,56 мм, соотношением C/D 0,9 и ВГД 33/20 мм рт. ст. С 36-го дня жизни потребовалась двусторонняя антиглаукоматозная операция5).
Системные признаки следующие:
Примерно у 50–60% пациентов с АРС развивается глаукома. Она часто возникает в детском или молодом возрасте, но может проявляться и в младенчестве повышением внутриглазного давления. Необходимы регулярные измерения внутриглазного давления и оценка зрительного нерва. Подробнее см. в разделе «Стандартное лечение».
АРС наследуется по аутосомно-доминантному типу с полной пенетрантностью. Однако даже в одной семье у людей с одинаковой генетической мутацией могут наблюдаться очень разные клинические проявления (вариабельная экспрессивность)1).
В крупном когортном исследовании мутации FOXC1 и PITX2 были связаны с широким спектром глаукомы от детства до взрослого возраста9). Случаи, первоначально диагностированные клинически как первичная врожденная глаукома (ПВГ), могут быть переклассифицированы с помощью генетического тестирования. Когда признаки переднего сегмента у младенцев неявны, генетическое тестирование может способствовать точной диагностике типа заболевания.
В случаях с микроделецией в области гена PITX2 перекрытие делеций NEUROG2, UGT8 и NDST4 может привести к задержке развития и умственной отсталости8)3).
Kawanami et al. (2023) сообщили о 3-летнем японском мальчике с микроделецией 2,5 Мb в 4q25 (включающей PITX2, NEUROG2 и ANK2). У него наблюдались пупочная грыжа, колобома радужки и задержка развития, но ЭКГ была нормальной, несмотря на делецию ANK2. Гаплонедостаточность NEUROG2 была предложена в качестве возможной причины задержки развития8).
Вследствие аутосомно-доминантного наследования вероятность передачи от родителя с мутацией составляет 50%. Пенетрантность полная, но фенотип варьирует: тяжесть симптомов может значительно различаться при одной и той же мутации1). Рекомендуется генетическое тестирование и генетическое консультирование.
Диагноз ARS основывается на двусторонних аномалиях угла передней камеры и радужки. Диагностическим условием является наличие заднего эмбриотоксона с частичным прикреплением периферической радужки13). Если задний эмбриотоксон не подтверждается при биомикроскопии, необходима гониоскопия. При наличии системных аномалий требуется полное педиатрическое обследование для выявления синдрома ARS13).
Обратите внимание, что легкий задний эмбриотоксон встречается у 8–15% нормальной популяции, но сам по себе не связан с глаукомой и т.д. Сбор семейного анамнеза также важен для диагностики.
В классификации детской глаукомы согласно Руководству по лечению глаукомы (5-е издание) ARS рассматривается как типичный пример глаукомы, связанной с врожденными аномалиями развития глаза13). Диагноз ставится, когда имеющиеся с рождения аномалии развития глаза соответствуют диагностическим критериям детской глаукомы.
Основные заболевания, которые следует дифференцировать с ARS, перечислены ниже.
| Заболевание | Отличие от ARS |
|---|---|
| Синдром ICE | Односторонний, приобретенный, преобладание у женщин |
| Аномалия Петерса | Центральное помутнение роговицы, дефект мембраны Десцемета |
| Аниридия | Роговичный паннус, гипоплазия фовеа |
| Задняя полиморфная дистрофия роговицы | Двусторонний, семейный, без половых различий |
Синдром ICE (прогрессирующая атрофия радужки, синдром Чендлера и др.) требует дифференциации от ARS, но основным отличительным признаком является то, что ICE является односторонним и приобретенным, тогда как ARS является двусторонним и врожденным.
В настоящее время не существует этиотропного лечения самого АРС. Основу терапии составляют контроль глаукомы и наблюдение за системными осложнениями. Тактика лечения соответствует таковой при ранней врожденной глаукоме (первичная врожденная глаукома: ПВГ) 13).
Глаукома осложняет примерно 50–60% случаев АРС. Медикаментозное лечение проводится по общим принципам лечения глаукомы, но часто неэффективно.
Препараты, снижающие продукцию водянистой влаги
Бета-блокаторы : один из препаратов первой линии. Безопасны и эффективны, но у детей часто неэффективны.
Ингибиторы карбоангидразы (ИКА) в виде глазных капель : бринзоламид и др. Возможно сочетание с бета-блокаторами.
Альфа-2-адреномиметики (бримонидин) : противопоказаны детям до 2 лет из-за нейропсихических эффектов (апноэ, брадикардия, гипотензия, мышечная гипотония, угнетение ЦНС) 13).
Препараты, усиливающие отток водянистой влаги
Аналоги простагландинов : латанопрост, травопрост и др. Считается, что у детей их эффективность ниже, чем у взрослых 13).
Пример : У 7-летнего мальчика долгосрочный контроль с помощью травопроста + бринзоламида 5). У 77-летнего мужчины ВГД оставалось 35 мм рт. ст., несмотря на латанопрост, тимолол и бринзоламид, что указывает на трудность контроля 2).
Согласно одному отчету, нет разницы в эффективности между агонистами FP-рецепторов простаноидов и бета-блокаторами 13).
У младенцев доза глазных капель относительно велика по сравнению с массой тела и площадью поверхности, поэтому следует по возможности использовать препараты с низкой концентрацией 13).
Если медикаментозное лечение не позволяет контролировать внутриглазное давление, проводится хирургическое вмешательство 10)13).
Сообщенные осложнения после GDD включают: мелкая передняя камера 13,6%, гипотония 11,7%, хориоидальный выпот 8,3%, эндофтальмит 1,7% 14).
Chakraborty et al. (2022) сообщили о случае отслойки сетчатки, связанной с синдромом Аксенфельда-Ригера (15-летний мальчик). Наблюдались микроофтальмия и подвывих хрусталика; после витрэктомии ВГД повысилось до 41 мм рт. ст., образовалась склеральная стафилома. Была проведена диодная циклофотокоагуляция, в итоге ВГД достигло 18 мм рт. ст. 7).
Частота успеха угловой хирургии ниже, чем при ПКГ 13). При трабекулэктомии с ММС долгосрочная частота успеха через 2 года составляет около 59%; при ГДУ – 87% через 12 месяцев и 77% через 24 месяца 14). В рефрактерных случаях может потребоваться несколько операций.
Основной причиной АРС является дефект миграции и дифференцировки клеток нервного гребня. Нарушение развития клеток нервного гребня в передней камере, углу переднего сегмента, лицевых костях, зубах, сердечно-сосудистой системе и коже вокруг пупка приводит к порокам развития нескольких органов.
В конце эмбрионального периода недифференцированные эндотелиальные клетки, покрывающие переднюю камеру, в норме исчезают из радужки и угла. При АРС этот процесс исчезновения нарушается, и недифференцированные эндотелиальные клетки остаются на радужке, образуя тяжи. В углу возникает высокое прикрепление радужки, механически покрывающее трабекулярную сеть.
Гистологически наблюдается аномальное распространение монослоя эндотелиоподобных клеток с десцеметоподобной мембраной от задней поверхности роговицы через переднюю камеру, угол и поверхность радужки. Мембрана присутствует в квадрантах с эктропионом сосудистой оболочки и девиацией зрачка, тогда как в противоположных квадрантах отмечается атрофия радужки.
FOXC1 и PITX2 являются факторами транскрипции, которые связываются с определенными последовательностями ДНК для регуляции экспрессии нижележащих генов. Оба действуют синергично в развитии переднего сегмента глаза и регулируют общие нижележащие гены-мишени 3). Форкхед-домен FOXC1 (ДНК-связывающий домен из 110 аминокислот) является функционально наиболее важным 2), и предполагается, что мутации в этом домене более сильно связаны с нейропсихиатрическими симптомами.
Указываются два механизма повышения внутриглазного давления.
Степень отсутствия радужки и количество отростков радужки в углу не обязательно коррелируют с тяжестью глаукомы. Однако высокая степень сращения радужки в углу предрасполагает к глаукоме.
Мутации FOXC1 в большей степени способствуют врожденной глаукоме по сравнению с другими мутациями 1), и морфологические аномалии цилиарного тела и угла оттока могут способствовать повышению ВГД 1).
У мышей с мутацией FOXC1 наблюдаются уменьшение и структурные аномалии коллагеновых волокон в строме роговицы, а также повреждение клеток стромы роговицы 1). Кроме того, FOXC1 действует как ингибитор неоваскуляризации роговицы (через контроль биодоступности VEGF) 1); потеря этого ингибирования из-за мутации FOXC1 приводит к неоваскуляризации роговицы.
FOXC1, как фактор транскрипции семейства FOX, также играет важную роль в развитии мозга 4).
Систематический обзор показал, что аномалии белого вещества возникают в 41,3% случаев АРС 4). Мутации FOXC1 могут индуцировать церебральную болезнь мелких сосудов (CSVD), гиперинтенсивность белого вещества, расширенные периваскулярные пространства, микрокровоизлияния и лакунарные инфаркты.
Ohkubo et al. (2025) подтвердили с помощью МРТ головного мозга у 2-летнего японского мальчика (мутация FOXC1: c.240del, p.Y81Ifs21) перивентрикулярные поражения белого вещества, расширенные периваскулярные пространства и извитость-расширение вертебробазилярной артерии. У отца был инфаркт мозга в возрасте 18 лет 4).
Обзор 95 случаев мутаций FOXC1 выявил нейропсихиатрические симптомы (трудности в обучении, эпилепсия, умственная отсталость, бред ревности и т.д.) в 6,3% случаев, а у 83,3% случаев с мутациями в forkhead-домене наблюдались нейропсихиатрические симптомы 2).
Да. Систематический обзор показывает, что аномалии белого вещества возникают примерно в 41% случаев АРС с мутацией FOXC1 4). Мутации FOXC1 могут быть связаны с риском церебральной болезни мелких сосудов и инсульта; поэтому важно долгосрочное неврологическое наблюдение, особенно при АРС, связанном с мутацией FOXC1.
В последние годы с помощью секвенирования нового поколения и полногеномного секвенирования было сообщено о многих новых мутациях.
Wowra et al. (2024) идентифицировали крупную делецию (новую мутацию), включающую часть экзона 1 FOXC1 и всю 3’UTR, при АРС у трех польских сестер. Фенотипы в одной семье сильно различались, и первоначально был ошибочно диагностирован синдром Чандлера 1).
Jiang et al. (2024) идентифицировали сложную геномную перестройку, включающую делецию 6,15 Мб хромосомы 4q25, содержащую PITX2, инверсию 45,71 Мб и делецию 14 п.н., в китайской семье с АРС 1 типа. У 11-летней девочки ВГД составляло 43,5/44,0 мм рт. ст. 3).
Другие сообщения о новых мутациях включают FOXC1 p.Phe136Leu (вилочный домен) 2), FOXC1 p.S82R (мутация de novo) 5) и FOXC1 c.240del, p.Y81Ifs21 4).
Yoshino et al. (2024) сообщили о случае 77-летнего японского мужчины с АРС 3 типа. С 72 лет появился бред ревности, была подтверждена лейкоэнцефалопатия. Обзор литературы 95 случаев мутаций FOXC1 показал, что у 6,3% (6/95) наблюдались нейропсихиатрические симптомы, из которых 83,3% (5/6) имели мутацию вилочного домена 2).
Это наблюдение предполагает, что функциональный домен мутации FOXC1 может быть вовлечен в проявление нейропсихиатрических симптомов, что указывает на важность долгосрочного наблюдения с точки зрения психического здоровья.
Было высказано предположение, что мутации FOXC1 могут индуцировать CSVD и увеличивать риск инсульта 4). Профилактика и раннее вмешательство при нейроваскулярных заболеваниях у пациентов с АРС считаются будущими направлениями исследований.
Понимание патологии «склерализации» роговицы, вызванной мутациями FOXC1, прогрессирует 1). Ожидается, что понимание молекулярных механизмов помутнения роговицы приведет к разработке новых методов лечения с использованием генной терапии, антифибротических препаратов и биоматериалов 1).