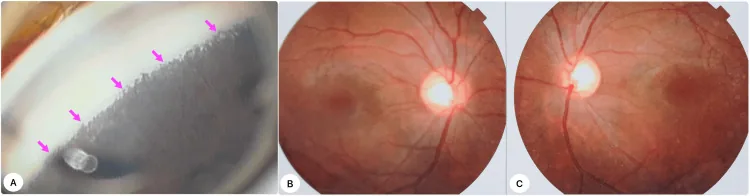
Iqbal MI, et al. A Landmark Case of Childhood Glaucoma Care in Bangladesh: Gonioscopy-Assisted Transluminal Trabeculotomy in Primary Congenital Glaucoma. Cureus. 2025. Figure 1. PMCID: PMC11934033. License: CC BY.
根据ANZRAG队列的基因诊断率报告,总体24.7%(125/506例)获得了分子诊断。在PCG中,30.4%(41/135例)可实现分子诊断,包括CYP1B1双等位基因突变15.6%(21例)、TEK杂合突变5.9%(8例)、CPAMD8 3.7%(5例)、FOXC1杂合突变3.7%(5例)。基于基因诊断的PCG亚型重新分类发生在10.4%的病例中(FOXC1突变重新分类为ARS,CPAMD8突变重新分类为ASD)7)。CYP1B1双等位基因突变在PCG女性中更常见(66.7% vs 33.3%,P=0.02)7)。
带引流管的植入物手术(GDD):用于滤过手术无效的病例。一项针对Ahmed瓣膜和Baerveldt植入物的32项研究、1221只眼的荟萃分析显示,术前平均眼压31.8±3.4 mmHg,术后12个月平均眼压降至16.5 mmHg(95% CI 15.517.6)8)。成功率随时间下降:12个月87%(95% CI 0.830.91),24个月77%(95% CI 0.71~0.83),120个月37%。Ahmed组和Baerveldt组的成功率无显著差异8),主要并发症包括前房变浅13.6%、低眼压11.7%、脉络膜渗出8.3%。
Pazos M, Traverso CE, Viswanathan A; European Glaucoma Society. European Glaucoma Society - Terminology and guidelines for glaucoma, 6th Edition. Br J Ophthalmol. 2025;109(Suppl 1):1-212. doi:10.1136/bjophthalmol-2025-egsguidelines. PMID:41026937.
Rao A. Histopathological changes in the trabecular meshwork in primary congenital glaucoma. American journal of ophthalmology case reports. 2025;38:102340. doi:10.1016/j.ajoc.2025.102340. PMID:40475128; PMCID:PMC12138567.
Song Y, Zhang X, Weinreb RN.. Gonioscopy-assisted transluminal trabeculotomy in primary congenital glaucoma. Am J Ophthalmol Case Rep. 2022;25:101366. doi:10.1016/j.ajoc.2022.101366. PMID:35146211; PMCID:PMC8818529.
Elhusseiny AM, Aboulhassan RM, El Sayed YM, Gawdat GI, Elhilali HM. Gonioscopy-Assisted Transluminal Trabeculotomy following Failed Glaucoma Surgery in Primary Congenital Glaucoma: One-Year Results. Case reports in ophthalmological medicine. 2023;2023:6761408. doi:10.1155/2023/6761408. PMID:37304219; PMCID:PMC10250098.
Dada T, Satpute K, Bukke AN, Verma S. Endoscope-assisted goniotomy in primary congenital glaucoma with corneal opacification. BMJ case reports. 2022;15(11). doi:10.1136/bcr-2022-252958. PMID:36357114; PMCID:PMC9660561.
Knight LSW, Ruddle JB, Taranath DA, et al. Childhood and early onset glaucoma classification and genetic profile in a large Australasian disease registry. Ophthalmology. 2024;131(1):62-73.
Stallworth JY, O’Brien KS, Han Y, Oatts JT. Efficacy of Ahmed and Baerveldt glaucoma drainage device implantation in the pediatric population: a systematic review and meta-analysis. J AAPOS. 2023;27(3):139.e1-139.e10.
Stingl JV, Ortolano LC, Azuara-Blanco A, Hoffmann EM. Systematic Review of Instruments for the Assessment of Patient-Reported Outcomes and Quality of Life in Patients with Childhood Glaucoma. Ophthalmology. Glaucoma. 2024;7(4):391-400. doi:10.1016/j.ogla.2024.02.009. PMID:38423388.