Tipo a prevalenza retinica esterna

Sindrome dei punti bianchi
Punti chiave a colpo d’occhio
Sezione intitolata “Punti chiave a colpo d’occhio”1. Cos’è la sindrome dei punti bianchi?
Sezione intitolata “1. Cos’è la sindrome dei punti bianchi?”La sindrome dei punti bianchi (white dot syndromes) è un concetto denominato da Gass nel 1977, che si riferisce a un gruppo di malattie infiammatorie caratterizzate da multiple lesioni maculate bianche o giallo-biancastre nel fondo oculare. La definizione delle malattie target varia a seconda dei ricercatori, ma attualmente è ampiamente utilizzata per indicare un gruppo di malattie infiammatorie idiopatiche non infettive che colpiscono principalmente la retina esterna, l’EPR, la coriocapillare e la coroide 1).
Secondo le linee guida per la diagnosi e il trattamento dell’uveite (Jpn J Ophthalmol 2019;123(6):635-696), le classificazioni dell’uveite posteriore includono APMPPE, MEWDS, PIC, coroidite multifocale, retinochoroidopatia di Birdshot, coroidite serpiginosa e AZOOR come entità distinte. Il termine generico «sindrome dei punti bianchi» viene utilizzato come concetto riassuntivo per queste condizioni2).
Concetto di classificazione
Sezione intitolata “Concetto di classificazione”Grazie ai recenti progressi nell’imaging multimodale (incluso OCT-A), la sindrome dei punti bianchi viene ora classificata in tre gruppi in base allo strato principale della lesione1).
Tipo a predominanza della coriocapillare
APMPPE・coroidite serpiginosa・PIC
- La vasculite obliterativa della coriocapillare è il nucleo della patologia
- L’OCT-A mostra un flow void della coriocapillare
- Le alterazioni della retina esterna e dell’EPR sono secondarie
Tipo a predominanza dello stroma coroidale
Coroidoretinopatia di Birdshot
- Infiltrazione linfocitaria dello stroma coroidale come lesione primaria
- All’OCT-A: flow void nello strato di Haller, iniziale risparmio dei coriocapillari
- Decorso cronico progressivo e forte associazione con HLA-A29
Inoltre, è stato proposto il concetto di complesso AZOOR, che integra MEWDS, AZOOR, PIC, MFC, AMN, AIBSE e AAOR come un continuum con una base genetica autoimmune/infiammatoria comune3).
Epidemiologia in Giappone
Sezione intitolata “Epidemiologia in Giappone”Secondo le statistiche della Società Giapponese di Infiammazione Oculare, la proporzione di ciascuna malattia della sindrome dei punti bianchi rispetto al totale delle uveiti è la seguente 2).
| Malattia | Proporzione sul totale delle uveiti |
|---|---|
| MEWDS | Circa 1-2% (rapporti nazionali) |
| APMPPE | Rara (nessuna statistica chiara sull’incidenza annuale) |
| PIC | Raro |
| Coroidite serpiginosa | Circa 0,3% |
| Retinocoroidite a pallini (birdshot) | Raro (più comune nei bianchi, estremamente raro in Giappone) |
| AZOOR | raro (numero di segnalazioni in aumento negli ultimi anni) |
2. Quadro clinico comune
Sezione intitolata “2. Quadro clinico comune”I sintomi di ciascuna malattia sono diversi, ma le seguenti caratteristiche cliniche sono comuni al gruppo delle sindromi dei punti bianchi 1, 2).
Schema comune dei sintomi soggettivi
Sezione intitolata “Schema comune dei sintomi soggettivi”- Riduzione dell’acuità visiva : l’entità varia da lieve (MEWDS, AZOOR) a grave (coroidite serpiginosa, CNV complicante PIC)
- Fotopsia : sintomo più comune che riflette un danno alla retina esterna e ai fotorecettori
- Scotoma e difetto del campo visivo : spesso scotoma paracentrale o centrale corrispondente alla lesione
- Metamorfopsia : si verifica in caso di lesione maculare o CNV
Presenza di infiammazione del segmento anteriore
Sezione intitolata “Presenza di infiammazione del segmento anteriore”| Gruppo di malattie | Infiammazione della camera anteriore e del vitreo |
|---|---|
| MEWDS, APMPPE, PIC | Di solito assente (APMPPE può essere lieve) |
| MFC (MFCwP) | Infiammazione della camera anteriore + vitreite (punto di differenziazione dalla PIC) |
| Birdshot | Infiammazione della camera anteriore assente o minima, vitreite presente |
| Coroidite serpiginosa | Infiammazione della camera anteriore e vitrite solitamente lievi |
| AZOOR | Solitamente assente |
3. Approccio alla diagnosi differenziale
Sezione intitolata “3. Approccio alla diagnosi differenziale”Età, sesso, unilaterale vs bilaterale, recidiva
Sezione intitolata “Età, sesso, unilaterale vs bilaterale, recidiva”Giovane donna, unilaterale, miglioramento spontaneo
Donne giovani e di mezza età, bilaterale, rischio di CNV
Giovane o di mezza età, bilaterale, esordio acuto
- Colpisce prevalentemente i 20-30enni (età media 25 anni), senza differenza di sesso
- Bilaterale, acuto, tendenza alla risoluzione spontanea
- Attenzione alla complicanza di vasculite cerebrale (esame urgente in caso di sintomi neurologici)
Età media/avanzata, bilaterale, cronico progressivo
Birdshot·Coroidite serpiginosa
- Birdshot: 40-60 anni, leggermente più frequente nelle donne
- A forma di serpente: 30-50 anni, leggermente più comune negli uomini
- Entrambe le malattie sono croniche, ricorrenti e richiedono immunosoppressione a lungo termine.
- Tasso di positività HLA-A29 nella malattia di Birdshot (caucasici): 80–98 %
Diagramma di flusso per la diagnosi differenziale
Sezione intitolata “Diagramma di flusso per la diagnosi differenziale”眼底に白点状病変 │ ├─ 片眼性? │ ├─ YES → MEWDS・AZOOR・AMN を考慮 │ │ ↳ FA で初期過蛍光 → MEWDS │ │ ↳ 眼底ほぼ正常・ERG 異常 → AZOOR │ └─ NO(両眼性) │ ├─ 急性発症・後極部大型白斑? │ └─ YES → APMPPE(FA 蛍光逆転現象を確認) │ ├─ 後極部小病変・近視女性・硝子体炎なし? │ └─ YES → PIC を考慮(CNV 検索:OCTA 必須) │ ├─ 小病変・硝子体炎あり・周辺部にも病変? │ └─ YES → MFC(MFCwP)を考慮 │ ├─ 乳頭周囲から蛇行状に進展・男性多め? │ └─ YES → 蛇行状脈絡膜炎(結核除外が最優先) │ └─ 後極部散弾状病変・中高年・HLA-A29? └─ YES → Birdshot 網脈絡膜症4. Confronto delle principali malattie
Sezione intitolata “4. Confronto delle principali malattie”Tabella comparativa principale: matrice delle 7 malattie
Sezione intitolata “Tabella comparativa principale: matrice delle 7 malattie”| Elemento | APMPPE | MEWDS | PIC | MFC (MFCwP) | Birdshot | Coroidite serpiginosa | AZOOR |
|---|---|---|---|---|---|---|---|
| Età di insorgenza | 20-30 anni (media 25 anni) | 20-50 anni | 18-40 anni (media 36 anni) | Media 30 anni | 40-60 anni | 30-50 anni | Principalmente intorno ai 35 anni |
| Sesso | Nessuna differenza di sesso | Predominanza femminile (1:4) | Predominanza femminile (circa 90%) | Predominanza femminile (più comune nelle donne bianche) | Leggermente più comune nelle donne | Leggermente più comune negli uomini | Predominanza femminile (circa 75%) |
| Monoculare/Bilaterale | Prevalentemente bilaterale | Monoculare (>95%) | Prevalentemente bilaterale (80%) | Bilaterale | Bilaterale | Bilaterale | Monoculare → bilaterale progressivo (alla fine 76% bilaterale) |
| Sintomi principali | Riduzione della vista, scotoma centrale, metamorfopsia | Riduzione della vista, fotopsia, visione offuscata | Scotoma, riduzione della vista, metamorfopsia | Miodesopsie, riduzione della vista, fotopsia | Riduzione della vista, emeralopia, discromatopsia | Riduzione della vista, scotoma paracentrale | Fotopsia, difetto del campo visivo (fondo oculare quasi normale) |
| Caratteristiche delle macchie bianche del fondo oculare | Macchie grandi color crema al polo posteriore (1/4–1/2 diametro del disco ottico) | Macchie multiple grigio-biancastre pallide dal polo posteriore all’equatore (100–200 μm) | Macchie piccole giallo-biancastre al polo posteriore (100–300 μm), 12–25 | Macchie grigio-giallastre dal polo posteriore alla periferia (45–350 μm), con vitreite | Macchie color crema a pallini da caccia dal polo posteriore all’equatore (1/4–1/2 diametro del disco ottico) | Lesioni grigio-giallastre geografiche che si estendono a serpentina dalla regione peripapillare | Fondo oculare quasi normale (fase acuta), atrofia degli strati esterni in fase tardiva |
| Reperti OCT | Disordine della zona ellissoidale + iperriflettività retinica esterna, atrofia parziale residua dopo recupero | Zona ellissoidale marcatamente disordinata/scomparsa (fase acuta) → recupero | Rialzo iperriflettente sotto RPE + rottura EZ (evoluzione in 5 stadi) | Iperriflettività sotto RPE + rottura EZ (simile a PIC) | Lesioni coroidali, edema maculare cistoide, scomparsa di EZ come prognosi sfavorevole | Fase attiva: iperriflettività retinica esterna + liquido sottoretinico. Fase cicatriziale: atrofia RPE | La scomparsa di EZ (IS/OS) è il reperto più importante (corrisponde ai difetti del campo visivo) |
| Reperti FA | Ipofluorescenza precoce → iperfluorescenza tardiva (fenomeno di inversione della fluorescenza) | Iperfluorescenza a ghirlanda (wreath-like) persistente dall’inizio | Fase attiva: iperfluorescenza precoce in fase arteriosa → perdita tardiva | Fase attiva: ipofluorescenza precoce → perdita tardiva | Iperfluorescenza della lesione (senza perdita) + perdita vascolare e iperfluorescenza papillare | Fase attiva: ipofluorescenza precoce → iperfluorescenza tardiva (perdita) | Di solito normale o anomalie minori |
| Risultati ICGA | Ipofluorescenza in tutte le fasi (riflette direttamente l’ischemia della coriocapillare) | Ipofluorescenza tardiva (più estesa dei punti bianchi) | Ipofluorescenza intermedia, utile per rilevare lesioni subcliniche | Ipofluorescenza precoce e in tutte le fasi | Ipofluorescenza precoce e intermedia → isofluorescenza tardiva (iniziale) → fase avanzata: ipofluorescenza in tutte le fasi | Ipofluorescenza in tutte le fasi (riflette un disturbo della circolazione coroidale) | Normalmente normale |
| Risultati FAF | Fase acuta: autofluorescenza bassa o eccessiva. Fase di remissione: autofluorescenza bassa | Fase acuta: autofluorescenza alta (iperfluorescenza prevalente). Dopo recupero: normalizzazione | Fase attiva: autofluorescenza bassa (ipoAF), alone iperautofluorescente al margine | Fase attiva: autofluorescenza bassa | Autofluorescenza bassa confluente peripapillare (presente nel 73% dei casi) | Fase attiva: margine iperautofluorescente + alone ipoautofluorescente. Fase quiescente: autofluorescenza bassa | Anomalie di autofluorescenza a banda da alta a bassa |
| Reperti OCT-A | Assenza di flusso coriocapillare (elevata concordanza con FA/ICGA) | Coriocapillare generalmente preservata (parziale assenza di flusso transitoria) | Assenza di flusso coriocapillare (sede di lesione infiammatoria) | Assenza di flusso coriocapillare | Assenza di flusso dello strato di Haller (iniziale) → stadio avanzato: assenza di flusso a tutto spessore | Assenza di flusso coriocapillare (grave) | Coriocapillare generalmente preservata |
| Ricorrente | Raro (sostanzialmente singolo episodio) | Recidiva in circa il 10% | Elevata (cronico ricorrente) | Elevata (episodi infiammatori ricorrenti) | Elevata (cronico con ricadute e remissioni) | Elevata (ripetizione ogni 3 mesi-4 anni) | Stabile entro 6 mesi nella maggior parte. In alcuni progressione |
| Tasso di CNV | raro | raro | 40–76% (alto rischio) | Fino al 60% | CNV sottoretinica: rara | Fino al 35% | Quasi assente |
| Associato a HLA | Nessuno | HLA-B51 (rapporto preliminare) | HLA-DR2 · HLA-DRB1*15 | Associato all’aplotipo IL-10 | HLA-A29 (80-98% nei caucasici) | HLA-B7 · HLA-A2 (rapporto di associazione) | Nessuno (predisposizione immunologica) |
| Strategia terapeutica | Osservazione (risoluzione spontanea), grave: steroidi | Osservazione, grave: steroidi a breve termine, CNV: anti-VEGF | Osservazione (senza CNV), CNV: anti-VEGF + steroidi, immunosoppressori | Steroidi + terapia immunomodulante, CNV: anti-VEGF | Steroidi + micofenolato mofetile / adalimumab (a lungo termine) | Steroidi + immunosoppressori (inclusi agenti alchilanti), CNV: anti-VEGF | Osservazione, grave: bolo di steroidi |
| Prognosi visiva | Buona (frequente risoluzione spontanea) | Buona (attenzione a recidive e CNV) | Alto rischio di prognosi sfavorevole in caso di CNV associata | Rischio di prognosi sfavorevole in caso di CNV ed edema maculare | Senza trattamento, il 16-22% ha un’acuità visiva ≤ 0,1 a 10 anni | Irreversibile se coinvolge la fovea, fino al 25% degli occhi ha un’acuità finale < 20/200 | Per lo più stabile. Sfavorevole in caso di progressione del danno agli strati esterni |
Q
Quale malattia della sindrome dei punti bianchi ha la prognosi visiva peggiore?
A
La coroidite serpiginosa e la retino-coroidopatia a pallini (birdshot) hanno la prognosi visiva peggiore. Nella coroidite serpiginosa, fino al 25% degli occhi raggiunge un’acuità visiva finale inferiore a 20/200, e nella retino-coroidopatia a pallini, senza trattamento, il 16-22% dei pazienti presenta un’acuità visiva pari o inferiore a 0,1 dopo 10 anni2, 4). PIC e MFC presentano un alto rischio di prognosi sfavorevole in caso di complicanza da CNV. APMPPE e MEWDS hanno una forte tendenza alla risoluzione spontanea e una prognosi favorevole.
Quadro clinico dettagliato di ciascuna malattia
Sezione intitolata “Quadro clinico dettagliato di ciascuna malattia”APMPPE (epiteliopatia pigmentaria placoida multifocale posteriore acuta)
Sezione intitolata “APMPPE (epiteliopatia pigmentaria placoida multifocale posteriore acuta)”L’APMPPE si manifesta prevalentemente tra i 20 e i 30 anni (media 25 anni), senza differenze di sesso. Si ritiene che sia causata da una vasculite occlusiva delle arteriole afferenti del letto capillare coroidale, e si sospetta che un’infezione virale possa essere un fattore scatenante1, 2).
Sintomi prodromici e decorso
- Circa la metà dei pazienti presenta sintomi simil-influenzali (influenza, virus EB, varicella, infezione da streptococco, ecc.).
- Nel polo posteriore di entrambi gli occhi compaiono multiple macchie bianche discoidi color crema di diametro compreso tra 1/4 e 1/2 del diametro papillare.
- Le macchie bianche iniziano a scomparire dal centro in pochi giorni e scompaiono in 7-12 giorni, lasciando una lieve depigmentazione.
- Di solito un singolo episodio si risolve spontaneamente (le recidive sono rare).
- La prognosi visiva è solitamente buona, ma può essere sfavorevole nei casi gravi o in quelli che evolvono in coroidite a carta geografica.
Complicanza specifica: vasculite cerebrale (MCAT)
APMPPE と中枢神経血管炎の合併(MCAT: multiple cerebral arterial thrombosis)は重篤な合併症であり、頭痛・発熱・神経症状が出現した場合は緊急で脳 MRI・MRA を施行する必要がある。脳血管炎合併例ではメチルプレドニゾロンパルス療法と神経内科連携が必要となる2)。
placoid chorioretinitis spectrum との関連
APMPPE は PPM(persistent placoid maculopathy)・RPC(relentless placoid chorioretinitis)とともに「placoid chorioretinitis spectrum」を形成し、脈絡膜毛細血管板虚血を共通病態基盤とする5)。
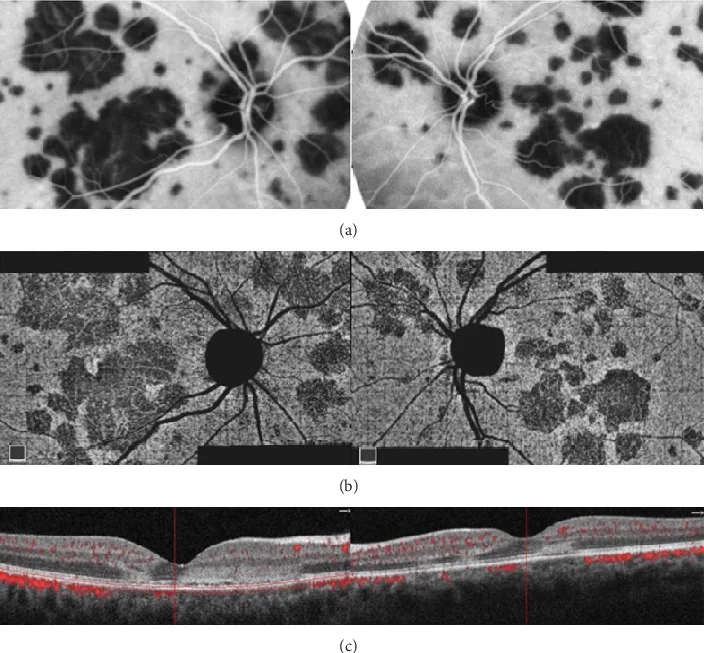
Oliveira MA, et al. Management of Acute Posterior Multifocal Placoid Pigment Epitheliopathy (APMPPE): Insights from Multimodal Imaging with OCTA. Case Rep Ophthalmol Med. 2020. Figure 5. PMCID: PMC7094199. License: CC BY.
両眼後極部に多発する地図状・斑状の病変がICGAでは低蛍光、OCTAでは脈絡毛細血管板の血流低下領域、OCTでは外網状層からRPEにかけての高反射変化として描出されている。本文「APMPPE(急性後部多発性斑状色素上皮症)」の項で扱うplacoid病変のマルチモーダル所見に対応する。
MEWDS (sindrome dei punti bianchi evanescenti multipli)
Sezione intitolata “MEWDS (sindrome dei punti bianchi evanescenti multipli)”La MEWDS si manifesta prevalentemente in donne di età compresa tra 20 e 50 anni (rapporto maschi:femmine 1:4) ed è caratterizzata da interessamento unilaterale, acuto e risoluzione spontanea.
Quadro clinico caratteristico
- Piccole macchie multiple grigio-biancastre pallide (100-200 μm) negli strati profondi della retina a livello dell’EPR, dal polo posteriore all’equatore
- foveal granularity (granularità foveale) : presente nel 74-96% dei pazienti, può essere l’unico reperto residuo dopo la scomparsa dei punti bianchi. Mostra un pattern caratteristico alla NIR-FAF (autofluorescenza nel vicino infrarosso)9)
- aspetto a punti arancioni (orange-dot appearance) : reperto caratteristico alla fotografia del fondo oculare e all’imaging del fondo in luce infrarossa
- Lesione avorio (ivory lesion): cambiamento bianco pallido e sfumato al polo posteriore del fondo oculare
- Sintomi simil-influenzali precedono circa il 50% dei casi
- L’incidenza annuale è di circa 0,22 casi per 100.000 persone, con recidiva nel 10% dei casi
Iperfluorescenza a ghirlanda (wreath-like) alla FA
L’iperfluorescenza a ghirlanda caratteristica già nelle fasi precoci della FA è un punto chiave per la diagnosi di MEWDS. Le lesioni a punti bianchi diventano iperfluorescenti precocemente alla FA e non mostrano espansione tardiva. Questa iperfluorescenza precoce è un importante punto di differenziazione dall’ipofluorescenza precoce dell’APMPPE (fenomeno di inversione della fluorescenza)1, 9).
Continuità con il complesso AZOOR
La MEWDS è principalmente dovuta a una distruzione transitoria della zona ellissoidale (linea IS/OS) dei fotorecettori ed è compresa come una malattia appartenente al complesso AZOOR. L’OCT mostra in fase acuta un’alterazione/scomparsa della zona ellissoidale, che migliora con il recupero visivo3).
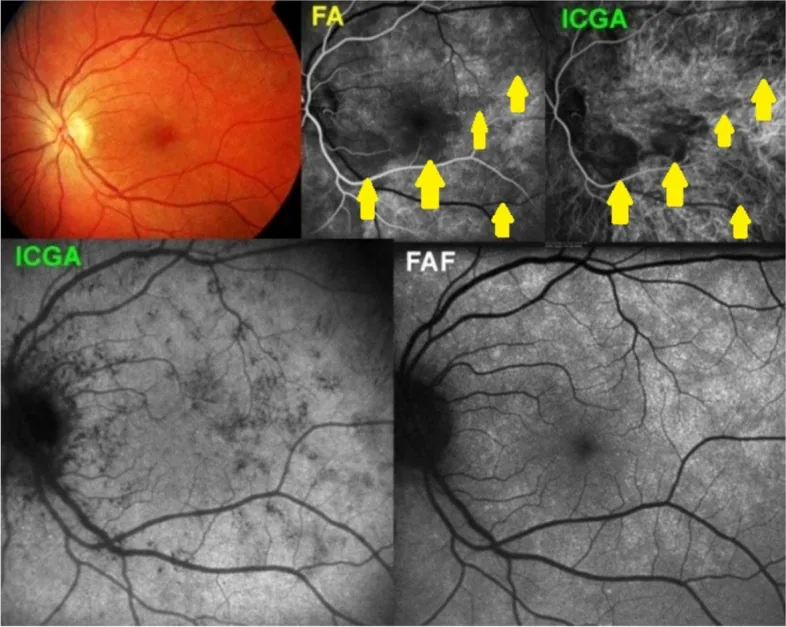
Papasavvas I, et al. Choroidal vasculitis as a biomarker of inflammation of the choroid. Indocyanine Green Angiography (ICGA) spearheading for diagnosis and follow-up, an imaging tutorial. J Ophthalmic Inflamm Infect. 2024. Figure 5. PMCID: PMC11618284. License: CC BY.
Lesioni puntiformi bianche pallide sono sparse al polo posteriore, con anomalie del flusso sanguigno puntiformi alla FA e all’ICGA, e corrispondente iperautofluorescenza alla FAF. Corrisponde ai reperti multimodali di iperfluorescenza a ghirlanda alla FA e punti ipofluorescenti all’ICGA trattati nella sezione «MEWDS (sindrome dei punti bianchi multipli evanescenti)» del testo.
PIC (Punctate Inner Choroidopathy)
Sezione intitolata “PIC (Punctate Inner Choroidopathy)”La PIC si verifica prevalentemente in giovani donne (circa 90%) di età compresa tra 18 e 40 anni con miopia (media circa -5 D).
Quadro clinico caratteristico
- Piccole macchie giallo-biancastre di 100-300 μm limitate al polo posteriore, di solito 12-25
- Assenza di infiammazione della camera anteriore e vitreite (punto chiave di differenziazione dalla MFC)
- Le lesioni attive sono visibili all’OCT come rilievi iperriflettenti sotto l’EPR
- La cicatrizzazione lascia piccole lesioni atrofiche
La complicanza da CNV (40-76%) è il principale problema clinico
La complicanza più importante della PIC è la CNV, con un tasso di complicanza riportato tra il 40 e il 76%7, 8). La CNV si verifica più facilmente a causa dei seguenti fattori:
- Fragilità della membrana di Bruch dovuta a assottigliamento coroidale miopico
- Distruzione della membrana di Bruch da infiammazione sotto l’RPE
- Aumento della produzione locale di citochine infiammatorie (VEGF, ecc.)
L’OCT-A si è dimostrato più sensibile dell’AG per lo screening della CNV, e si raccomanda un monitoraggio regolare con OCT-A. Un improvviso peggioramento della metamorfopsia è un segno di sviluppo di CNV e richiede un esame tempestivo.
Associazione con malattie sistemiche
È stata riportata un’associazione tra PIC e sarcoidosi; nei pazienti con lesioni polmonari multiple, devono essere eseguiti TC torace, ACE sierico e lisozima. È stata anche riportata un’associazione con HLA-DR2 e HLA-DRB1*153).
MFC (coroidite multifocale, MFCwP)
Sezione intitolata “MFC (coroidite multifocale, MFCwP)”La MFC (coroidite multifocale con panuveite; MFCwP) è una malattia dello stesso spettro della PIC, ma il principale punto di differenziazione è la presenza di vitrite e infiammazione della camera anteriore7).
Quadro clinico caratteristico
- Multiple macchie giallo-grigiastre di 45–350 μm compaiono non solo al polo posteriore ma anche nella periferia media.
- Decorso cronico recidivante (episodi infiammatori ricorrenti) caratteristico
- Alta frequenza di complicanza da membrana epiretinica (ERM) (fino al 35%), che influenza la prognosi visiva a lungo termine
- In alcuni casi l’infiammazione non può essere controllata senza terapia immunosoppressiva
Avvertenze terapeutiche
La MFC tende a non risolversi spontaneamente e spesso richiede una terapia immunomodulante a lungo termine. Quando i soli steroidi sono insufficienti, si utilizzano metotrexato (MTX), azatioprina (AZA) o micofenolato mofetile (MMF). In caso di complicanza da CNV, è importante un approccio bidirezionale con terapia anti-VEGF e terapia immunomodulante7, 8).
Coroidopatia a pallini (Birdshot chorioretinopathy)
Sezione intitolata “Coroidopatia a pallini (Birdshot chorioretinopathy)”La Birdshot si manifesta in persone di mezza età (40-60 anni, media 50 anni), con una leggera prevalenza femminile (1,5:1). È più comune nei caucasici ed è una delle associazioni genetiche più forti conosciute con HLA-A29 (rischio relativo 50-224 volte nei caucasici)4).
Reperti caratteristici del fundus
- Macchie color crema (da 1/4 a 1/2 diametro del disco ottico) simili a segni di pallini, simmetricamente bilaterali dal polo posteriore all’equatore.
- Le macchie evolvono in lesioni cicatriziali senza pigmentazione.
- Possono associarsi vasculite retinica e edema della papilla.
Alterazioni funzionali caratteristiche
- Cecità notturna e discromatopsia : compaiono precocemente, talvolta prima del calo visivo
- ERG totale di tipo negativo : presente precocemente, con progressione l’ampiezza dell’onda a diminuisce
- Ritardo dell’ERG flicker a 30 Hz : indicatore più sensibile per il monitoraggio dell’attività, rileva anomalie prima del calo visivo17)
Avvertenze per i pazienti giapponesi
Poiché la frequenza di HLA-A29 nei giapponesi è bassa, la sensibilità diagnostica di HLA-A29 è limitata. La diagnosi deve basarsi sui reperti clinici dei criteri di classificazione SUN 2021 (lesioni a pallini di fucile al fundus, lieve infiammazione del segmento anteriore, presenza di vitrite)10).
Complicanze a lungo termine
- Edema maculare cistoide (CME): causa principale del calo visivo
- Edema papillare e atrofia ottica
- Con l’uso di impianti steroidei (fluocinolone), si osserva un aumento della pressione intraoculare fino al 40% dei pazienti, che può richiedere una trabeculectomia
Coroidopatia serpiginosa (Serpiginous choroidopathy)
Sezione intitolata “Coroidopatia serpiginosa (Serpiginous choroidopathy)”La coroidite serpiginosa è una coroidite cronica bilaterale che si verifica tra i 30 e i 50 anni (leggermente più frequente nei maschi), caratterizzata da lesioni grigio-gialle a carta geografica che si estendono in modo serpiginoso dalla regione peripapillare.
Pattern di estensione caratteristico
- Inizia centripetamente intorno alla papilla ottica (peripapillare), con il bordo della lesione che si espande gradualmente in modo serpiginoso
- Fase attiva: comparsa di un orlo grigio-biancastro al bordo della lesione
- Fase cicatriziale: fissazione come lesione atrofica corioretinica
- In caso di recidiva, una nuova infiammazione compare sempre al bordo della cicatrice preesistente (questo è caratteristico)
- L’intervallo tra le recidive varia da 3 mesi a 4 anni, con grande variabilità individuale.
Il più importante: differenziazione dal tipo correlato alla tubercolosi (SLC)
La coroidite serpiginosa-like tubercolare (serpiginous-like choroiditis; SLC) è molto simile alla coroidite serpiginosa all’imaging, ma la strategia terapeutica è fondamentalmente diversa:
| Punto di differenziazione | Coroidite serpiginosa | Tipo correlato alla tubercolosi (SLC) |
|---|---|---|
| Distribuzione delle lesioni | Centrata intorno alla papilla, centripeta | Polo posteriore alla periferia, multiple |
| IGRA/TST | Negativo | Positivo |
| Forma della lesione | A carta geografica, continua | Multiple piccole lesioni discontinue |
| Trattamento | Steroidi + immunosoppressori | Farmaci antitubercolari obbligatori |
L’uso di immunosoppressori nella SLC peggiora notevolmente la tubercolosi, quindi il test IGRA (Quantiferon) prima del trattamento è assolutamente indispensabile 2).
Gestione della complicanza CNV (fino al 35%)
Nella coroidite serpiginosa, la CNV si verifica fino al 35% dei casi e causa perdita irreversibile della vista se coinvolge la fovea. Le iniezioni intravitreali di anti-VEGF (bevacizumab, ranibizumab) sono efficaci 18).
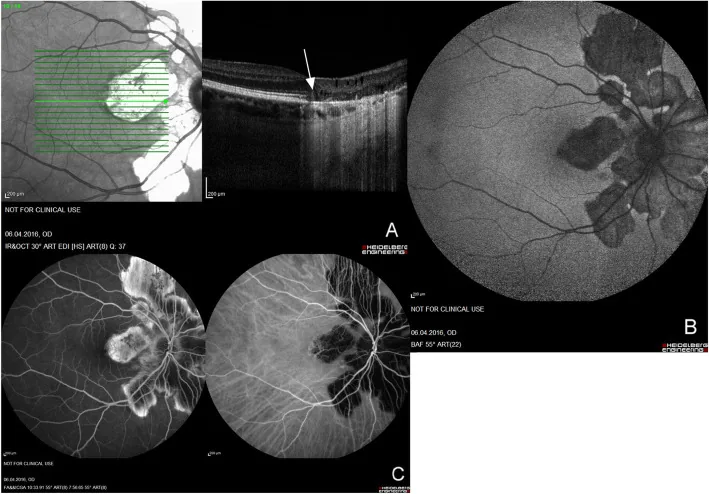
Macedo S, et al. Optical coherence tomography angiography (OCTA) findings in Serpiginous Choroiditis. BMC Ophthalmol. 2020. Figure 1. PMCID: PMC7325353. License: CC BY.
L’autofluorescenza del fondo, l’angiografia con fluoresceina e l’OCT mostrano lesioni corioretiniche serpiginose che si estendono centrifugamente dal disco ottico. Ciò corrisponde al pattern di estensione serpiginosa peripapillare discusso nella sezione «Coroidite serpiginosa».
AZOOR (retinopatia esterna occulta zonale acuta)
Sezione intitolata “AZOOR (retinopatia esterna occulta zonale acuta)”L’AZOOR è un concetto di malattia proposto da Gass nel 1992. Si tratta di una retinopatia esterna che causa improvvisa perdita della vista, difetti del campo visivo e fotopsia, nonostante un fondo oculare quasi normale3).
Concetto del complesso AZOOR
Il complesso AZOOR, proposto da Jampol et al., è un concetto che comprende MEWDS, AZOOR, PIC, MFC, AMN (neuroretinopatia maculare acuta), AIBSE e AAOR come un continuum con una base genetica autoimmune/infiammatoria comune3).
Caratteristiche cliniche specifiche
- Si verifica prevalentemente in giovani donne miopi di età compresa tra 20 e 50 anni.
- Fotopsia (visione di luci) compare spesso all’esordio (in particolare luci a forma di banda o arco)
- Inizia monolateralmente e diventa bilaterale nel 76% dei casi
- Il fondo oculare è quasi normale nella fase acuta (caratteristico è il divario tra riduzione dell’acuità visiva e reperti del fondo)
- I difetti del campo visivo hanno un pattern a bande irregolari (spesso contigui alla macchia cieca)
- Possono essere associate malattie autoimmuni (tiroidite di Hashimoto, sclerosi multipla)
L’OCT e l’ERG sono la chiave per la diagnosi
- La scomparsa o offuscamento della zona ellissoidale (linea IS/OS) all’OCT è il reperto più importante.
- Il recupero funzionale delle aree in cui lo strato esterno è scomparso all’OCT non è prevedibile (utile anche per la prognosi).
- Una riduzione dell’ampiezza all’ERG multifocale può essere rilevata anche in presenza di fondo oculare normale (l’ERG multifocale è più sensibile dell’ERG a campo totale).
- La FAF infrarossa può talvolta delineare il confine tra la lesione e l’area normale.
Trattamento e prognosi
Non esiste una terapia consolidata per l’AZOOR. I casi lievi possono essere solo osservati, ma i casi gravi (riduzione dell’acuità visiva o difetto esteso del campo visivo) richiedono terapia pulsata con metilprednisolone (1.000 mg × 3 giorni) seguita da prednisolone orale2). La maggior parte dei casi si stabilizza entro 6 mesi, ma il campo visivo non si recupera nelle aree con danno residuo dello strato esterno.
Q
Come si differenzia l'AZOOR dalla neurite ottica retrobulbare?
A
L’AZOOR e la neurite ottica retrobulbare richiedono una differenziazione poiché entrambi presentano un fondo oculare quasi normale con riduzione dell’acuità visiva e del campo visivo. I punti chiave per la differenziazione sono: ① nell’AZOOR si ha una riduzione dell’ampiezza dell’ERG multifocale, mentre nella neurite ottica retrobulbare l’ERG è normale; ② nell’AZOOR i difetti del campo visivo mostrano un pattern irregolare a nastro o ad arco, mentre nella neurite ottica retrobulbare è comune uno scotoma centrale; ③ nell’AZOOR il RAPD è solitamente lieve, mentre nella neurite ottica retrobulbare si osserva un RAPD marcato; ④ all’OCT, l’AZOOR mostra la scomparsa della zona ellissoidale, mentre la neurite ottica retrobulbare mostra edema papillare o assottigliamento della RNFL, utile per la differenziazione2, 3).
5. Utilizzo degli esami di imaging
Sezione intitolata “5. Utilizzo degli esami di imaging”Suddivisione dei ruoli per modalità
Sezione intitolata “Suddivisione dei ruoli per modalità”Comprendere chiaramente il ruolo di ciascun esame di imaging è essenziale per una diagnosi accurata e una valutazione dell’attività della sindrome dei punti bianchi1, 5).
| Modalità | Indicazione/ruolo più forte |
|---|---|
| AGF (angiografia con fluoresceina) | Valutazione della perdita dai vasi retinici, dall’EPR e dalla coriocapillare. Conferma del fenomeno di inversione della fluorescenza nell’APMPPE. Conferma dell’iperfluorescenza a ghirlanda nel MEWDS. Valutazione della vasculite (Birdshot). |
| ICGA (angiografia con verde indocianina) | Valutazione diretta dei disturbi della circolazione coroidale. Rilevamento delle lesioni più precocemente rispetto all’AGF (in particolare Birdshot, APMPPE, serpiginosa). Rilevamento di lesioni più estese rispetto ai reperti clinici (MEWDS, PIC). La più sensibile per rilevare lesioni attive. |
| FAF (autofluorescenza del fondo) | Valutazione non invasiva del danno all’RPE. Determinazione dell’attività (APMPPE, serpiginosa). Diagnosi di MEWDS (punti bianchi precoci iperautofluorescenti). Valutazione della cronicità in Birdshot (ipoautofluorescenza peripapillare 73%) |
| OCT (tomografia a coerenza ottica) | Valutazione della zona ellissoidale (reperti diagnostici in MEWDS e AZOOR). Valutazione dell’evoluzione delle lesioni in 5 stadi (PIC). Valutazione di CNV ed edema maculare. Prognosi (perdita di EZ → prognosi visiva sfavorevole) |
| OCT-A | Rilevamento non invasivo di aree di non perfusione coriocapillare (APMPPE, serpiginosa, PIC). Rilevamento precoce e sensibile di CNV (più sensibile della FA in PIC e MFC). Valutazione del flusso sanguigno coroidale per strato in Birdshot. Monitoraggio dell’efficacia terapeutica |
| ERG multifocale / ERG a campo totale | Diagnosi di AZOOR (riduzione dell’ampiezza dell’ERG anche con fondo quasi normale). Monitoraggio dell’attività della Birdshot (ritardo del flicker a 30 Hz più sensibile). Valutazione della risposta al trattamento. |
Guida « quale esame per primo » per patologia
Sezione intitolata “Guida « quale esame per primo » per patologia”Quando si sospetta MEWDS
Quando si sospetta APMPPE
Quando si sospetta PIC/MFC
Quando si sospetta una coroidite serpiginosa
Q
Quale eseguire per primo tra FA e ICGA?
A
La strategia varia a seconda della malattia. Nell’APMPPE e nella coroidite serpiginosa, l’ICGA visualizza più direttamente i disturbi della circolazione coroidale, quindi eseguirla contemporaneamente o dopo la FA approfondisce la comprensione della patologia. Nel MEWDS, l’iperfluorescenza a ghirlanda alla FA è importante per la diagnosi. Tuttavia, essendo esami invasivi, l’OCT-A fornisce ora molte informazioni alternative e la combinazione FAF e OCT-A viene utilizzata come valutazione iniziale1, 5).
Significato delle lesioni ipofluorescenti all’ICGA
Sezione intitolata “Significato delle lesioni ipofluorescenti all’ICGA”L’ipofluorescenza delle lesioni della sindrome dei punti bianchi all’ICGA è il riflesso diretto di un’interruzione del flusso sanguigno coroidale (occlusione dei coriocapillari)1). L’ICGA è più sensibile della FA come modalità per valutare la circolazione coroidale e presenta le seguenti caratteristiche:
- Ipofluorescenza totale: osservata in APMPPE, coroidite serpiginosa, PIC e MFC. Riflette un’occlusione grave dell’ischemia del letto capillare coroidale.
- Ipofluorescenza tardiva (FA normale) : L’ipofluorescenza tardiva all’ICGA nella MEWDS è spiegata da un’alterata captazione dell’ICG dovuta a un’anomalia dell’EPR, non a un coinvolgimento della coriocapillare (la coriocapillare è generalmente risparmiata all’OCT-A)1).
- Flow void dello strato di Haller → flow void a tutto spessore in fase avanzata : Pattern di progressione in due fasi caratteristico della Birdshot, che inizia nello stroma coroidale e si estende alla coriocapillare14).
L’ICGA è anche superiore alla FA e all’OCT-A nel rilevare lesioni subcliniche clinicamente invisibili, e in particolare nella MEWDS e nella PIC mostra lesioni coroidali più estese dei punti bianchi1, 15).
Mappa delle malattie secondo i pattern di ipo e iperfluorescenza alla FAF
Sezione intitolata “Mappa delle malattie secondo i pattern di ipo e iperfluorescenza alla FAF”I pattern della FAF (autofluorescenza del fondo) riflettono lo stato metabolico dell’EPR e sono utili per la diagnosi e la valutazione dell’attività delle sindromi dei punti bianchi16).
| Pattern FAF | Malattia/stadio | Significato |
|---|---|---|
| Iperautofluorescenza (iper AF) | MEWDS fase acuta · bordo attivo di Serpiginosa | Accumulo di prodotti di degenerazione dei fotorecettori (A2E, ecc.) nell’RPE |
| Ipoautofluorescenza (ipo AF) | APMPPE fase cicatriziale · lesione attiva PIC · peripapillare Birdshot | Scomparsa/perdita di funzione dell’RPE |
| Ipo-AF centrale con alone iper-AF periferico | Bordo attivo serpiginoso, PIC | Pattern di danno RPE al bordo attivo |
| AF anomalo a banda | AZOOR | Corrisponde alla distribuzione del danno dello strato esterno dei fotorecettori |
| IperAF granulare foveale | MEWDS (NIR-FAF) | Visualizzazione non invasiva della granularità foveale |
Nel Birdshot, l’ipoAF confluente peripapillare è presente nel 73% dei casi, utile come indicatore di cronicità17).
Pattern di difetto della zona ellissoidale (EZ) all’OCT
Sezione intitolata “Pattern di difetto della zona ellissoidale (EZ) all’OCT”La valutazione della EZ (zona ellissoidale, ex linea IS/OS) svolge un ruolo centrale nell’attività e nella prognosi della sindrome dei punti bianchi1).
| Riscontro EZ | Malattia/stadio | Prognosi |
|---|---|---|
| EZ marcatamente disorganizzato → recupero | MEWDS fase acuta → fase di recupero | Buono (recupero EZ e recupero visivo correlati) |
| Scomparsa dell’EZ (corrispondente alla lesione) | Fase attiva di AZOOR | Nessun recupero funzionale dell’area scomparsa |
| Disorganizzazione dell’EZ + iperriflettività della retina esterna | Fase acuta di APMPPE | Atrofia parziale residua dopo il recupero |
| Rialzo iperriflettente sotto RPE + rottura della zona ellissoidale | PIC·MFC (evoluzione in 5 stadi) | Prognosi sfavorevole in caso di CNV associata |
| Scomparsa della zona ellissoidale (con edema maculare cistoide) | Birdshot in fase avanzata | Fattore di rischio per prognosi visiva sfavorevole |
Il choriocapillaris flow void all’OCT-A mostra un’elevata concordanza con i reperti di FA e ICGA (particolarmente utile nell’APMPPE e nella coroidite serpiginosa) 5, 13).
Pattern di fluorescenza alla FA: ritardo di riempimento vs iperfluorescenza vs perdita
Sezione intitolata “Pattern di fluorescenza alla FA: ritardo di riempimento vs iperfluorescenza vs perdita”| Pattern FA | Malattia | Significato clinico |
|---|---|---|
| Ipofluorescenza precoce → iperfluorescenza tardiva (fenomeno di inversione della fluorescenza) | APMPPE | Ischemia della coriocapillare. Difetto di riempimento precoce → perdita tardiva di colorante dai tessuti circostanti. |
| Iperfluorescenza precoce a corona (wreath-like) | MEWDS | Riflette direttamente il danno a RPE/fotorecettori. Non si espande in fase tardiva, punto chiave per la diagnosi differenziale. |
| Ipofluorescenza precoce → perdita tardiva | Serpiginosa / MFC in fase attiva | Evidenza di coroidite attiva |
| Iperfluorescenza precoce arteriosa → perdita tardiva | Fase attiva di PIC | Sospetta presenza di CNV infiammatoria |
| Perdita vascolare + iperfluorescenza papillare (senza perdita) | Birdshot | Prova diretta di vasculite retinica |
| Normale o lieve | AZOOR | Caratterizzato da dissociazione: difetti del campo visivo e anomalie ERG nonostante FA negativa |
6. Principi generali di trattamento
Sezione intitolata “6. Principi generali di trattamento”Scelta della strategia terapeutica
Sezione intitolata “Scelta della strategia terapeutica”Il trattamento della sindrome dei punti bianchi varia notevolmente in base alla storia naturale della malattia, alla gravità e alla presenza o meno di CNV.
Osservazione (nessun trattamento necessario) come principio
Sezione intitolata “Osservazione (nessun trattamento necessario) come principio”- Forte tendenza alla risoluzione spontanea, spesso senza trattamento specifico2)
- MEWDS: scomparsa dei punti bianchi e recupero visivo in poche settimane. Steroidi orali solo in caso di gravità o edema papillare associato.
- APMPPE: regressione delle macchie bianche in 7-12 giorni, prognosi visiva solitamente buona.
Malattie in cui la terapia steroidea è il trattamento principale
Sezione intitolata “Malattie in cui la terapia steroidea è il trattamento principale”APMPPE (grave, con papillite) · PIC (lesione attiva vicino alla fovea) · AZOOR (casi gravi)
- Iniziare con prednisolone 30–60 mg/die e poi ridurre gradualmente
- In caso di vasculite cerebrale associata ad APMPPE, è necessaria la terapia pulsata con metilprednisolone e la collaborazione con il neurologo
- Criterio di gravità per AZOOR: acuità visiva corretta dell’occhio migliore < 0,3 (linee guida diagnostiche della Società Giapponese di Oftalmologia)
Malattie che richiedono una terapia immunosoppressiva a lungo termine
Sezione intitolata “Malattie che richiedono una terapia immunosoppressiva a lungo termine”Coroidite a pallini (Birdshot), coroidite serpiginosa, MFC
Birdshot (gestione a lungo termine)
- Prednisolone 0,5–1 mg/kg/die all’inizio
- Micofenolato mofetile (MMF) 2–3 g/die (immunosoppressore di prima linea)
- Metotrexato (MTX) 10–25 mg/settimana
- Azatioprina (AZA) 1–3 mg/kg/die
- Casi refrattari: adalimumab (93,2% usato come farmaco biologico di prima scelta)
- Senza trattamento, il 16–22% ha un’acuità visiva ≤ 0,1 in 10 anni
Coroidite serpiginosa (gestione a lungo termine)
- Prednisolone 40–80 mg/die all’inizio (riduzione graduale)
- Azatioprina 1–2,5 mg/kg/die (terapia di mantenimento di prima linea)
- Micofenolato mofetile / metotrexato (alternativa)
- Casi refrattari: clorambucile (il più potente; ≤0,2 mg/kg/die, emocromo settimanale obbligatorio)
- Farmaci biologici: adalimumab (91,0% lo raccomandano come prima scelta)
- Escludere la tubercolosi (se IGRA positivo, somministrare prima antitubercolari) e poi introdurre l’immunosoppressore
Terapia immunomodulante della MFC
Sezione intitolata “Terapia immunomodulante della MFC”La MFC ha un decorso cronico recidivante, pertanto dopo la riduzione graduale degli steroidi è spesso necessaria una terapia di mantenimento con immunomodulatori.
| Farmaco | Dose indicativa | Note |
|---|---|---|
| Metotrexato (MTX) | 10–25 mg/settimana | Associare acido folico 1 mg/die. Monitoraggio epatotossicità |
| Azatioprina (AZA) | 1–3 mg/kg/die | Raccomandata verifica attività TPMT. Attenzione alla mielosoppressione |
| Micofenolato mofetile (MMF) | 1–3 g/giorno | I sintomi gastrointestinali sono effetti collaterali comuni |
| Ciclosporina (CsA) | 3–5 mg/kg/giorno | Monitoraggio obbligatorio della funzionalità renale e della pressione arteriosa |
| Adalimumab | 40 mg/2 settimane (sottocute) | Casi refrattari o steroido-dipendenti. Screening per tubercolosi obbligatorio |
Gestione della CNV associata
Sezione intitolata “Gestione della CNV associata”La CNV è la complicanza più importante che influenza la prognosi visiva, ed è particolarmente frequente nella PIC, nella MFC e nella coroidite serpiginosa.
| Malattia | Tasso di CNV associata | Trattamento |
|---|---|---|
| PIC | 40–76% | Iniezione intravitreale di anti-VEGF (bevacizumab, ranibizumab, aflibercept) + steroidi. Strategia OCTA PRN. |
| MFC | Fino al 60% | Iniezione intravitreale di anti-VEGF + terapia immunomodulante |
| Coroidite serpiginosa | Fino al 35% | Iniezione intravitreale di anti-VEGF (bevacizumab, ranibizumab) |
| MEWDS·APMPPE | Raro | Anti-VEGF (se CNV confermata) |
| Birdshot | Raro | Immunosoppressione sistemica + anti-VEGF in caso di CNV |
Q
La sola terapia anti-VEGF è sufficiente per la CNV?
A
La CNV infiammatoria (iCNVM) differisce dalla CNV della degenerazione maculare senile; il controllo dell’infiammazione di base è importante anche per prevenire la recidiva della CNV. Nella PIC e nella MFC, un approccio bidirezionale con anti-VEGF e steroidi (o immunosoppressori) è considerato efficace; il solo anti-VEGF lascia un rischio di recidiva 7, 8).
Precauzioni terapeutiche
Sezione intitolata “Precauzioni terapeutiche”| Punto di attenzione | Malattia interessata |
|---|---|
| Escludere la tubercolosi in via prioritaria (prima dell’immunosoppressione) | Coroidite serpiginosa · MFC |
| Considerare HLA-A29 (diagnosi) | Birdshot (bassa sensibilità nei giapponesi) |
| Rischio di aumento della pressione intraoculare con impianto steroideo | Birdshot (fino al 40% necessita di trabeculectomia) |
| Rischio di mielosoppressione e tumori maligni con clorambucile | Coroidite serpiginosa (necessario esame del sangue settimanale) |
| Riacutizzazione dopo infezione o vaccinazione da COVID-19 | PIC・MEWDS (segnalazione di recidiva) |
| Risposta di emergenza in caso di vasculite cerebrale | APMPPE (cefalea, sintomi neurologici → RM encefalo urgente) |
Q
Quali esami devono essere eseguiti per primi quando si usano immunosoppressori?
A
Prima di iniziare un immunosoppressore per la sindrome dei punti bianchi, gli esami obbligatori da eseguire sono i seguenti: ① esclusione della tubercolosi tramite IGRA (Quantiferon) (massima priorità nella coroidite serpiginosa), ② screening per epatite virale con antigene HBs, anticorpi HBc e anticorpi HCV (prevenzione della riattivazione sotto immunosoppressione), ③ radiografia del torace e TC (esclusione di tubercolosi e sarcoidosi), ④ emocromo completo e test di funzionalità epatica e renale (verifica dei valori basali). Per i farmaci biologici come l’adalimumab, lo screening per la tubercolosi è obbligatorio anche secondo il foglio illustrativo19, 20).
Q
Come si determinano il numero e il programma di somministrazione della terapia anti-VEGF?
A
A differenza della degenerazione maculare legata all’età, la CNV infiammatoria (iCNVM) può regredire spontaneamente quando l’infiammazione è controllata. Di solito si utilizza una strategia PRN (al bisogno), con iniezione ogni volta che una CNV attiva viene confermata all’OCT-A. Può essere somministrata una dose di carico iniziale (3 iniezioni consecutive), ma in combinazione con la terapia immunosoppressiva il numero di iniezioni necessarie può essere ridotto. Con il solo anti-VEGF il rischio di recidiva è elevato, quindi è importante un approccio bidirezionale con il controllo dell’infiammazione di base 7, 8).
6.5 Correlazione con malattie sistemiche
Sezione intitolata “6.5 Correlazione con malattie sistemiche”Ciascuna malattia della sindrome dei punti bianchi è associata a specifiche malattie sistemiche o infezioni, pertanto è importante un’esclusione sistematica prima del trattamento.
| Malattia | Malattie/condizioni sistemiche associate | Significato clinico |
|---|---|---|
| APMPPE | Vasculite cerebrale (MCAT) · Infezione da streptococco · Virus EB | Cefalea · Sintomi neurologici → RM encefalo urgente |
| Birdshot | HLA-A29 (80–98% nei caucasici) · Simile alla sarcoidosi | Test HLA come ausilio diagnostico (bassa sensibilità nei giapponesi) |
| Coroidite serpiginosa | Tubercolosi (SLC) · HLA-B7/A2 | IGRA positivo → prima terapia antitubercolare |
| PIC | Sarcoidosi · HLA-DRB1*15 | Considerare TC torace e dosaggio ACE |
| MFC | Aplotipo IL-10·EBV·sarcoidosi | rivalutazione sistemica nei casi di recidiva cronica |
| MEWDS | Infezione da COVID-19·post-vaccinazione·HLA-B51 | L’infezione da SARS-CoV-2 funge da trigger immunitario |
| AZOOR | Tiroidite di Hashimoto, sclerosi multipla, malattie autoimmuni | Considerare esami della funzione tiroidea e autoanticorpi |
7. Punti particolari e conoscenze recenti per ciascuna malattia
Sezione intitolata “7. Punti particolari e conoscenze recenti per ciascuna malattia”APMPPE e spettro della corioretinite placoid
Sezione intitolata “APMPPE e spettro della corioretinite placoid”L’APMPPE è ora compresa in modo integrato come parte dello «spettro della corioretinite placoidale» insieme alla maculopatia placoidale persistente (PPM) e alla corioretinite placoidale incessante (RPC). In queste tre malattie, l’OCT-A mostra un pattern comune di flow void della coriocapillare, indicando che l’ischemia della coriocapillare è un substrato patologico comune5).
Klufas et al. (2017) hanno riportato che l’OCT-A rileva i flow void coriocapillari con un alto tasso di concordanza con FA e ICGA in tre malattie: APMPPE, PPM e RPC, supportando il concetto di spettro della corioretinite placoid5).
MEWDS e granularità foveale
Sezione intitolata “MEWDS e granularità foveale”La granularità foveale è un segno diagnostico riscontrato nel 74-96% dei casi di MEWDS e può rimanere l’unico reperto dopo la scomparsa dei punti bianchi. La FAF nel vicino infrarosso (NIR-FAF) mostra un caratteristico pattern granulare foveale9).
Spettro delle malattie di PIC e MFC
Sezione intitolata “Spettro delle malattie di PIC e MFC”PIC e MFC (MFCwP) condividono un background genetico comune (aplotype IL-10, HLA-DRB1*15) e sono considerati fenotipi diversi dello stesso spettro di malattie. I principali punti di differenziazione sono la presenza o assenza di vitrite e infiammazione della camera anteriore, nonché la distribuzione delle lesioni1, 3).
| Punto di differenziazione | PIC | MFC (MFCwP) |
|---|---|---|
| Vitrite | Assente | Presente (importante punto di differenziazione) |
| Infiammazione della camera anteriore | Assente | Lieve |
| Distribuzione delle lesioni | Limitata al polo posteriore | Polo posteriore + periferia media |
| Dimensione della lesione | 100–300 μm | 45–350 μm |
| Tasso di concomitanza di CNV | 40–76% | Fino al 60% |
Birdshot e HLA-A29
Sezione intitolata “Birdshot e HLA-A29”L’associazione tra Birdshot e HLA-A29 è una delle più forti associazioni genetiche conosciute in tutte le malattie, con un rischio relativo aumentato da 50 a 224 volte nei pazienti bianchi4). Tuttavia, nei giapponesi, la bassa prevalenza di portatori di HLA-A29 limita la sensibilità della positività all’HLA-A29 nella diagnosi. È importante basarsi sui reperti clinici dei criteri di classificazione SUN 2021 (reperti del fundus, minima infiammazione della camera anteriore, vitreite) per la diagnosi10).
Complesso AZOOR e trigger virale
Sezione intitolata “Complesso AZOOR e trigger virale”Si ritiene che l’AZOOR si sviluppi quando a una predisposizione genetica (aplotipo IL-10, ecc.) si aggiunge un trigger ambientale (infezione virale, vaccino, farmaco, ecc.) e viene compreso come parte del complesso AZOOR insieme a MEWDS, PIC, AMN e AIBSE3). I casi di MEWDS dopo infezione da COVID-19 o vaccinazione stanno aumentando a livello globale, suggerendo che SARS-CoV-2 potrebbe agire come trigger immunitario11).
Q
La sindrome dei punti bianchi può verificarsi dopo un'infezione da COVID-19 o la vaccinazione?
A
Sì. Sono stati riportati diversi casi di insorgenza o recidiva di MEWDS, PIC e coroidite serpiginosa dopo infezione da COVID-19, suggerendo che l’infezione da SARS-CoV-2 possa fungere da trigger immunitario11, 12). Per il MEWDS è stata condotta una revisione sistematica di 27 casi dopo vaccinazione anti-COVID-19, con il vaccino a mRNA (Pfizer-BioNTech) come il più frequente. I soggetti con precedenti anamnestici dovrebbero consultare un oculista per il follow-up prima e dopo la vaccinazione.
Q
Qual è l'ordine di priorità degli esami da eseguire al primo sospetto di sindrome dei punti bianchi?
A
L’ordine di priorità raccomandato per gli esami in caso di primo sospetto di sindrome dei punti bianchi è il seguente: ① FAF + OCT (non invasivo, consente la valutazione iniziale di quasi tutte le malattie; visualizzazione dell’iper-AF nel MEWDS, valutazione della zona ellissoidale, sollevamento sotto l’EPR nel PIC) → ② OCT-A (rilevamento precoce di CNV, valutazione del flow void coriocapillare) → ③ FA + ICGA (se necessaria una diagnosi definitiva o una valutazione dell’attività). In caso di sospetto di coroidite serpiginosa, IGRA (esclusione della tubercolosi) deve essere prioritario prima della FA. In caso di sospetto di AZOOR, l’ERG (ERG multifocale) è obbligatorio1, 2).
8. Articoli correlati
Sezione intitolata “8. Articoli correlati”
急性後部多発性斑状色素上皮症(APMPPE)
両眼後極部に多発する斑状白斑と蛍光逆転現象が特徴。中枢神経血管炎合併に注意。
多発消失性白点症候群(MEWDS)
近視若年女性に好発する片眼性急性炎症。foveal granularity が診断的所見。
点状内層脈絡膜症(PIC)
若年近視女性に好発。CNV を40〜76%に合併し、OCTA による定期スクリーニングが必須。
多巣性脈絡膜炎(MFC)
硝子体炎を伴う PIC スペクトラム疾患。CNV を最大60%に合併する。
バードショット網脈絡膜症
HLA-A29 関連の慢性後部ぶどう膜炎。長期免疫抑制療法と ERG モニタリングが必須。
蛇行状脈絡膜炎
乳頭周囲から蛇行状に進展する慢性脈絡膜炎。結核との鑑別が最重要課題。
急性帯状潜在性網膜外層症(AZOOR)
眼底ほぼ正常にもかかわらず急性視野欠損・光視症をきたす外網膜症。ERG 振幅低下が診断の鍵。
特発性多巣性脈絡膜炎(IMC)
MFC と連続性を持つ特発性脈絡膜炎のスペクトラム記事。
9. Riferimenti
Sezione intitolata “9. Riferimenti”- Testi I, Modugno RL, Pavesio C. Multimodal imaging supporting the pathophysiology of white dot syndromes. Journal of ophthalmic inflammation and infection. 2021;11(1):32. doi:10.1186/s12348-021-00261-3. PMID:34529201; PMCID:PMC8446150.
- 日本眼炎症学会ぶどう膜炎診療ガイドライン作成委員会. ぶどう膜炎診療ガイドライン. 日眼会誌. 2019;123(6):635-696.
- Jampol LM, Becker KG. White spot syndromes of the retina: a hypothesis based on the common genetic hypothesis of autoimmune/inflammatory disease. American journal of ophthalmology. 2003;135(3):376-9. doi:10.1016/s0002-9394(02)02088-3. PMID:12614757.
- Agrawal R, et al. The role of HLA-A29 in birdshot chorioretinopathy and immune checkpoint inhibitor-related uveitis. Am J Ophthalmol. 2025. doi:10.1016/j.ajo.2024.01.007
- Klufas MA, Phasukkijwatana N, Iafe NA, Prasad PS, Agarwal A, Gupta V, et al. Optical Coherence Tomography Angiography Reveals Choriocapillaris Flow Reduction in Placoid Chorioretinitis. Ophthalmology. Retina. 2017;1(1):77-91. doi:10.1016/j.oret.2016.08.008. PMID:31047399.
- Stattin M, Forster J, Ahmed D, Krepler K, Ansari-Shahrezaei S. Swept Source-Optical Coherence Tomography Angiography for Management of Secondary Choroidal Neovascularization in Punctate Inner Choroidopathy. Case reports in ophthalmology. 2021;12(1):232-238. doi:10.1159/000511669. PMID:33976688; PMCID:PMC8077444.
- Spaide RF, Goldberg N, Freund KB. Redefining multifocal choroiditis and panuveitis and punctate inner choroidopathy through multimodal imaging. Retina (Philadelphia, Pa.). 2013;33(7):1315-24. doi:10.1097/IAE.0b013e318286cc77. PMID:23584703.
- Leclaire MD, Clemens CR, Eter N, Mihailovic N. Choroidale Neovaskularisation infolge einer “punctate inner choroidopathy”, dargestellt mittels optischer Kohärenztomographie-Angiographie. Ophthalmologe. 2021;118:842-846. doi:10.1007/s00347-020-01200-8.
- Mantovani A, Invernizzi A, Staurenghi G, Herbort CP.. Multiple Evanescent White Dot Syndrome: A Multimodal Imaging Study of Foveal Granularity. Ocul Immunol Inflamm. 2019;27(1):141-147. doi:10.1080/09273948.2017.1353104. PMID:28981397.
- Standardization of Uveitis Nomenclature (SUN) Working Group. Classification Criteria for Birdshot Chorioretinitis. American journal of ophthalmology. 2021;228:65-71. doi:10.1016/j.ajo.2021.03.059. PMID:33845003; PMCID:PMC8517033.
- Chen N, Mandell M, Arjmand P. Multimodal imaging findings of multiple evanescent white dot syndrome in COVID-19 patients. IDCases. 2024;38:e02110. doi:10.1016/j.idcr.2024.e02110. PMID:39582747; PMCID:PMC11585666.
- Seddigh S, Pinto A, Zaki AM, Gupta RR. Serpiginous Choroiditis After COVID-19 Infection. Journal of vitreoretinal diseases. 2025;9(2):246-252. doi:10.1177/24741264241297936. PMID:39554631; PMCID:PMC11562243.
- Pakzad-Vaezi K, Khaksari K, Chu Z, Van Gelder RN, Wang RK, Pepple KL.. Swept-Source OCT Angiography of Serpiginous Choroiditis. Ophthalmol Retina. 2018;2(7):712-719. doi:10.1016/j.oret.2017.11.001. PMID:30148243; PMCID:PMC6103638.
- Pepple KL, Chu Z, Weinstein J, Munk MR, Van Gelder RN, Wang RK. Use of en face swept-source optical coherence tomography angiography in identifying choroidal flow voids in 3 patients with birdshot chorioretinopathy. JAMA Ophthalmol. 2018;136(11):1288-1292. doi:10.1001/jamaophthalmol.2018.3474. PMID:30128478. PMCID:PMC6248174
- Khochtali S, Dridi T, Abroug N, Ksiaa I, Lupidi M, Khairallah M. Swept-Source Optical Coherence Tomography Angiography Shows Choriocapillaris Flow Reduction in Multiple Evanescent White Dot Syndrome. Journal of current ophthalmology. 2020;32(2):211-215. doi:10.4103/JOCO.JOCO_107_20. PMID:32671309; PMCID:PMC7337020.
- Yeh S, Forooghian F, Wong WT, Faia LJ, Cukras C, Lew JC, Wroblewski K, Weichel ED, Meyerle CB, Sen HN, Chew EY, Nussenblatt RB.. Fundus autofluorescence imaging of the white dot syndromes. Arch Ophthalmol. 2010;128(1):46-56. doi:10.1001/archophthalmol.2009.368. PMID:20065216; PMCID:PMC3025103.
- Minos E, Barry RJ, Southworth S, et al. Birdshot chorioretinopathy: current knowledge and new concepts in pathophysiology, diagnosis, monitoring and treatment. Orphanet J Rare Dis. 2016;11:61. doi:10.1186/s13023-016-0429-8. PMID:27175923; PMCID:PMC4866419.
- Maleki A, Maldonado Cerda A, Garcia CM, et al. Chlorambucil combination therapy in refractory serpiginous choroiditis: a cure? Am J Ophthalmol Case Rep. 2021;21:101014. doi:10.1016/j.ajoc.2021.101014. PMID:33615036; PMCID:PMC7881218.
- Niederer RL, Al-Janabi A, Engelbrecht C, et al. Immunomodulatory therapy prescribing practices for non-infectious uveitis: a survey of international experts. Br J Ophthalmol. 2024;108:482-489.
- Tomkins-Netzer O, et al. Treatment of non-infectious uveitis with biologics: a survey of the International Ocular Inflammation Society. Br J Ophthalmol. 2022;106:482-488.