Das White-Dot-Syndrom (White-Dot-Syndrome) ist ein von Gass 1977 geprägter Begriff, der eine Gruppe entzündlicher Erkrankungen bezeichnet, die durch multiple weiße oder gelblich-weiße fleckige Läsionen im Augenhintergrund gekennzeichnet sind. Die Definition der betroffenen Erkrankungen variiert je nach Forscher, wird aber heute allgemein für eine Gruppe nichtinfektiöser idiopathischer entzündlicher Erkrankungen verwendet, die hauptsächlich die äußere Netzhaut, das RPE, die Choriokapillaris und die Aderhaut betreffen 1).
Gemäß den Leitlinien zur Diagnose und Behandlung von Uveitis (Jpn J Ophthalmol 2019;123(6):635-696) werden als Klassifikationen der hinteren UveitisAPMPPE, MEWDS, PIC, multifokale Choroiditis, Birdshot-Retinochoroidopathie, serpiginöse Choroiditis und AZOOR als eigenständige Erkrankungen aufgeführt. Der Oberbegriff „White-Dot-Syndrom“ wird als zusammenfassendes Konzept für diese Entitäten verwendet2).
Dank der Fortschritte in der multimodalen Bildgebung (einschließlich OCT-A) in den letzten Jahren wird das White-Dot-Syndrom nun anhand der hauptsächlich betroffenen Schicht in die folgenden drei Gruppen eingeteilt1).
Lymphozyteninfiltration des Aderhautstromas als Primärläsion
Im OCT-A Flow Void in der Haller-Schicht, initiale Schonung der Choriokapillaris
Chronisch progredienter Verlauf und starke Assoziation mit HLA-A29
Darüber hinaus wurde das Konzept des AZOOR-Komplexes vorgeschlagen, das MEWDS, AZOOR, PIC, MFC, AMN, AIBSE und AAOR als ein Kontinuum mit gemeinsamer genetischer Autoimmun-/Entzündungsbasis integriert3).
Laut Statistik der Japanischen Gesellschaft für Augenentzündung beträgt der Anteil der einzelnen Erkrankungen des Weißpunkt-Syndroms an der Gesamtzahl der Uveitiden wie folgt 2).
Die Symptome der einzelnen Erkrankungen unterscheiden sich, aber die folgenden klinischen Merkmale sind der Gruppe der White-Dot-Syndrome gemeinsam 1, 2).
Hohes Risiko einer schlechten Prognose bei CNV-Beteiligung
Risiko einer schlechten Prognose bei CNV und Makulaödem
Ohne Behandlung haben nach 10 Jahren 16–22 % eine Sehschärfe ≤ 0,1
Irreversibel bei Beteiligung der Fovea, bei bis zu 25 % der Augen endgültige Sehschärfe < 20/200
Meist stabil. Schlecht bei fortschreitender Schädigung der äußeren Schichten
QWelche Erkrankung des White-Dot-Syndroms hat die schlechteste Sehprognose?
A
Die serpiginöse Choroiditis und die Birdshot-Retinochoroidopathie haben die schlechteste Sehprognose. Bei der serpiginösen Choroiditis erreicht in bis zu 25% der Augen der endgültige Visus weniger als 20/200, und bei der Birdshot-Retinochoroidopathie sinkt der Visus ohne Behandlung bei 16–22% der Patienten innerhalb von 10 Jahren auf 0,1 oder weniger2, 4). PIC und MFC haben bei Komplikation durch CNV ein hohes Risiko für einen schlechten Verlauf. APMPPE und MEWDS zeigen eine starke Tendenz zur spontanen Besserung und haben eine gute Prognose.
Die APMPPE tritt bevorzugt im Alter von 20–30 Jahren (Durchschnitt 25 Jahre) auf, ohne Geschlechtsunterschied. Als Ursache wird eine obliterative Vaskulitis der zuführenden Arteriolen der chorioidalen Kapillarschicht angenommen, und eine Virusinfektion wird als Auslöser vermutet1, 2).
Prodromalsymptome und Verlauf
Etwa die Hälfte der Patienten zeigt grippeähnliche Symptome (Influenza, EB-Virus, Windpocken, Streptokokkeninfektion usw.).
Im hinteren Pol beider Augen treten multiple cremefarbene, scheibenförmige weiße Flecken von 1/4 bis 1/2 Papillendurchmesser auf.
Die weißen Flecken beginnen innerhalb weniger Tage von der Mitte aus zu verschwinden und verschwinden nach 7–12 Tagen, wobei eine leichte Depigmentierung zurückbleibt.
In der Regel heilt eine einzelne Episode spontan aus (Rückfälle sind selten).
Die Sehprognose ist in der Regel gut, kann aber bei schweren Fällen oder bei Übergang in eine landkartenartige Chorioiditis schlecht sein.
MEWDS tritt bevorzugt bei Frauen im Alter von 20–50 Jahren auf (Geschlechterverhältnis 1:4) und ist gekennzeichnet durch einseitiges, akutes Auftreten und spontane Rückbildung.
Charakteristisches klinisches Bild
Blasse, grau-weiße multiple kleine Flecken (100-200 μm) in den tiefen Netzhautschichten bis zur RPE-Ebene, vom hinteren Pol bis zur Äquatorregion
foveal granularity (foveale Granularität) : Bei 74–96 % der Patienten vorhanden, kann sie nach dem Verschwinden der weißen Punkte der einzige verbleibende Befund sein. Zeigt ein charakteristisches Muster in der Nahinfrarot-FAF (NIR-FAF)9)
Orange-Punkt-Erscheinung (orange-dot appearance) : charakteristischer Befund in Fundusfotografie und Nahinfrarot-Fundusbildgebung
Elfenbeinläsion (ivory lesion): blasse, verschwommene weiße Veränderung am hinteren Augenpol
Bei etwa 50 % der Fälle treten grippeähnliche Prodromi auf
Die jährliche Inzidenz beträgt etwa 0,22 Fälle pro 100.000 Personen, bei 10 % kommt es zu einem Rezidiv
Kranzförmige (wreath-like) Hyperfluoreszenz in der FA
Die charakteristische kranzförmige Hyperfluoreszenz bereits in der frühen FA ist ein Schlüsselpunkt für die Diagnose des MEWDS. Die weißen Punktläsionen werden in der frühen FA hyperfluoreszent und zeigen keine späte Ausdehnung. Diese frühe Hyperfluoreszenz ist ein wichtiges Unterscheidungsmerkmal zur frühen Hypofluoreszenz der APMPPE (Fluoreszenzinversionsphänomen)1, 9).
MEWDS ist hauptsächlich durch eine vorübergehende Zerstörung der Ellipsoidzone (IS/OS-Linie) der Photorezeptoren gekennzeichnet und wird als eine zum AZOOR-Komplex gehörende Erkrankung verstanden. Im OCT zeigt sich in der akuten Phase eine Störung/Verlust der Ellipsoidzone, die sich mit der Erholung des Sehvermögens bessert3).
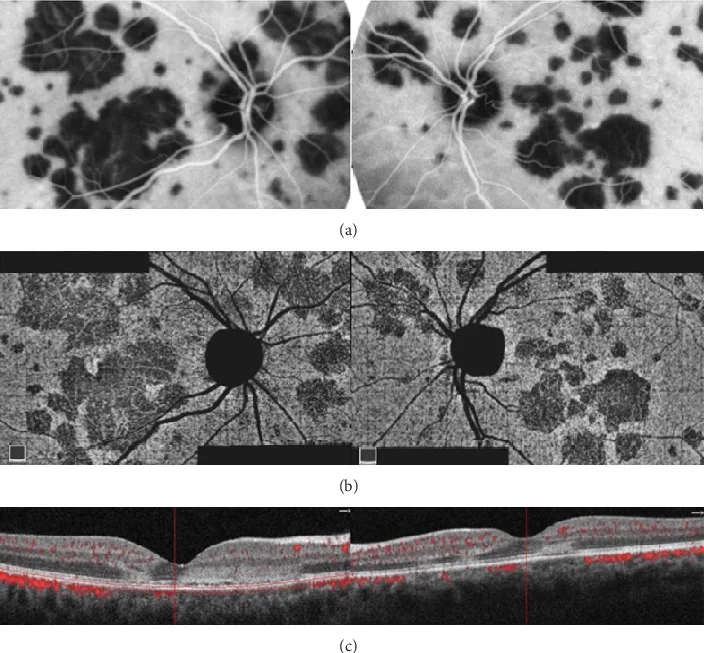
Papasavvas I, et al. Choroidal vasculitis as a biomarker of inflammation of the choroid. Indocyanine Green Angiography (ICGA) spearheading for diagnosis and follow-up, an imaging tutorial. J Ophthalmic Inflamm Infect. 2024. Figure 5. PMCID: PMC11618284. License: CC BY.
Am hinteren Pol sind blasse weiße punktförmige Läsionen verstreut, in FA und ICGA zeigen sich punktförmige Durchblutungsstörungen, in FAF korrespondierende Hyperautofluoreszenz. Entspricht den multimodalen Befunden von FA-wreath-artiger Hyperfluoreszenz und ICGA-Hypofluoreszenzpunkten, die im Abschnitt «MEWDS (multiples evaneszentes weißes Punktsyndrom)» des Textes behandelt werden.
PIC tritt vor allem bei jungen Frauen (ca. 90 %) im Alter von 18–40 Jahren mit Myopie (durchschnittlich etwa -5 D) auf.
Charakteristisches klinisches Bild
Auf den hinteren Pol beschränkte 100–300 μm große gelb-weiße Flecken, meist 12–25
Keine Vorderkammerentzündung oder Glaskörperentzündung (wichtigster Unterschied zur MFC)
Aktive Läsionen sind im OCT als hyperreflektive Erhebungen unter dem RPE sichtbar
Narbenbildung hinterlässt kleine atrophische Läsionen
CNV-Komplikation (40–76 %) ist das größte klinische Problem
Die wichtigste Komplikation der PIC ist die CNV, deren Komplikationsrate mit 40–76 % angegeben wird7, 8). Die CNV tritt leichter aufgrund folgender Faktoren auf:
Brüchigkeit der Bruch-Membran durch myopische Aderhautverdünnung
Zerstörung der Bruch-Membran durch Entzündung unter dem RPE
Erhöhte lokale Produktion von Entzündungszytokinen (VEGF usw.)
Die OCT-A hat sich als sensitiver als die FA für das CNV-Screening erwiesen, und eine regelmäßige OCT-A-Überwachung wird empfohlen. Eine plötzliche Verschlechterung der Metamorphopsie ist ein Zeichen für eine CNV-Entwicklung und erfordert eine sofortige Abklärung.
Assoziation mit systemischen Erkrankungen
Es wurde über ein gleichzeitiges Auftreten von PIC und Sarkoidose berichtet; bei Patienten mit multiplen Lungenläsionen sollten eine CT-Thorax, Serum-ACE und Lysozym untersucht werden. Auch eine Assoziation mit HLA-DR2 und HLA-DRB1*15 wurde berichtet3).
Die MFC (multifokale Choroiditis mit Panuveitis; MFCwP) ist eine Erkrankung desselben Spektrums wie die PIC, aber das wichtigste Unterscheidungsmerkmal ist das Vorhandensein von Vitritis und Vorderkammerentzündung7).
Charakteristisches klinisches Bild
Multiple gelb-graue Flecken von 45–350 μm treten nicht nur am hinteren Pol, sondern auch in der mittleren Peripherie auf.
Hohe Komplikationsrate der epiretinalen Membran (ERM) (bis zu 35 %), die die langfristige Sehprognose beeinflusst
In einigen Fällen kann die Entzündung ohne Immunsuppression nicht kontrolliert werden
Behandlungshinweise
Die MFC neigt nicht zur spontanen Rückbildung und erfordert oft eine langfristige immunmodulatorische Therapie. Wenn Steroide allein nicht ausreichen, werden Methotrexat (MTX), Azathioprin (AZA) oder Mycophenolatmofetil (MMF) eingesetzt. Bei Komplikation durch CNV ist ein bidirektionaler Ansatz mit Anti-VEGF-Therapie und immunmodulatorischer Therapie wichtig7, 8).
Birdshot tritt bei Personen mittleren bis höheren Alters (40–60 Jahre, Durchschnitt 50 Jahre) auf, mit einer leichten weiblichen Prädominanz (1,5:1). Sie ist häufiger bei Kaukasiern und stellt eine der stärksten bekannten genetischen Assoziationen mit HLA-A29 dar (relatives Risiko 50- bis 224-fach bei Kaukasiern)4).
Charakteristische Fundusbefunde
Cremefarbene Flecken (1/4 bis 1/2 Papillendurchmesser) in Schrotschussmuster, die symmetrisch beidseits vom hinteren Pol bis zum Äquator auftreten.
Die Flecken entwickeln sich zu Narbenläsionen ohne Pigmentierung.
Eine retinale Vaskulitis und Papillenschwellung können begleitend auftreten.
Charakteristische Funktionsveränderungen
Nachtblindheit und Farbsehstörungen : treten früh auf, manchmal vor der Sehverschlechterung
Negativer Ganzfeld-ERG-Typ : früh nachweisbar, mit fortschreitender Erkrankung nimmt die a-Wellen-Amplitude ab
Verzögerung des 30-Hz-Flicker-ERG : empfindlichster Indikator für die Aktivitätsüberwachung, erkennt Anomalien vor der Sehverschlechterung17)
Hinweise für japanische Patienten
Da die HLA-A29-Frequenz bei Japanern niedrig ist, ist die diagnostische Sensitivität von HLA-A29 eingeschränkt. Die Diagnose sollte auf den klinischen Befunden der SUN-2021-Klassifikationskriterien (Schrotschussläsionen im Fundus, geringe Vorderkammerentzündung, Vorhandensein einer Vitritis) basieren10).
Bei Verwendung von Steroidimplantaten (Fluocinolon) tritt bei bis zu 40 % der Patienten ein erhöhter Augeninnendruck auf, der eine Trabekulektomie erforderlich machen kann
Die serpiginöse Choroiditis ist eine bilaterale chronische Aderhautentzündung im Alter von 30–50 Jahren (Männer etwas häufiger), gekennzeichnet durch landkartenartige grau-gelbe Läsionen, die sich von der Papillenumgebung aus schlangenförmig ausbreiten.
Charakteristisches Ausbreitungsmuster
Beginnt zentripetal um die Sehnervenpapille (peripapillär) und breitet sich allmählich schlangenförmig am Läsionsrand aus
Aktive Phase: Auftreten eines grau-weißen Randsaums am Läsionsrand
Narbenphase: Fixierung als chorioretinale Atrophieläsion
Bei Rezidiven tritt eine neue Entzündung immer am Rand der bestehenden Narbe auf (dies ist charakteristisch)
Das Rezidivintervall variiert stark von 3 Monaten bis zu 4 Jahren.
Am wichtigsten: Abgrenzung zum tuberkuloseassoziierten Typ (SLC)
Die tuberkulöse serpiginöse Choroiditis (serpiginous-like choroiditis; SLC) ähnelt der serpiginösen Choroiditis in der Bildgebung, unterscheidet sich jedoch grundlegend in der Therapie:
Da die Anwendung von Immunsuppressiva bei SLC die Tuberkulose deutlich verschlimmert, ist ein IGRA-Test (Quantiferon) vor der Behandlung absolut unverzichtbar 2).
Bei der Serpiginösen Choroidopathie tritt in bis zu 35% der Fälle eine CNV auf, die bei Beteiligung der Fovea zu irreversiblen Sehverlust führt. Intravitreale Injektionen von Anti-VEGF (Bevacizumab, Ranibizumab) sind wirksam 18).
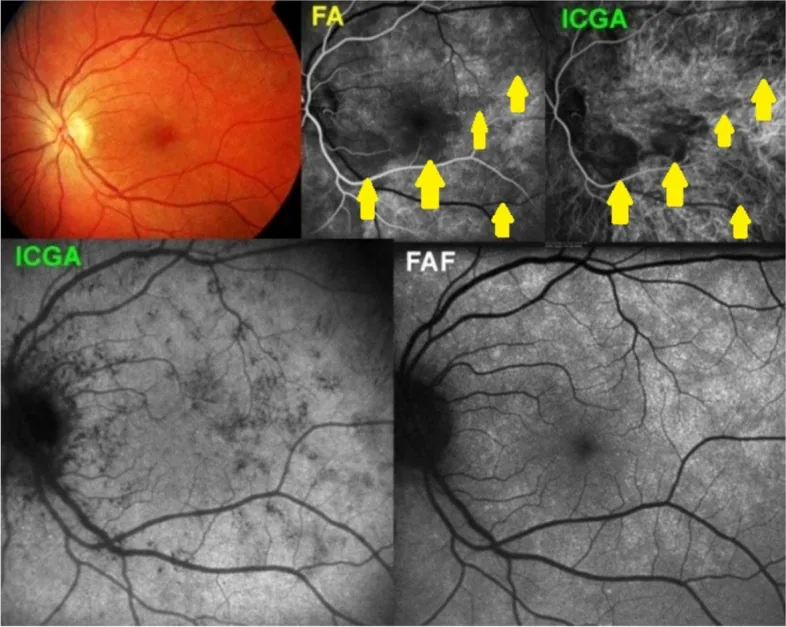
Macedo S, et al. Optical coherence tomography angiography (OCTA) findings in Serpiginous Choroiditis. BMC Ophthalmol. 2020. Figure 1. PMCID: PMC7325353. License: CC BY.
Fundusautofluoreszenz, Fluoreszenzangiographie und OCT zeigen serpiginöse chorioretinale Läsionen, die sich von der Papille aus zentrifugal ausbreiten. Dies entspricht dem im Abschnitt „Serpiginöse Choroidopathie“ beschriebenen peripapillären serpiginösen Ausbreitungsmuster.
AZOOR ist ein von Gass 1992 vorgeschlagenes Krankheitskonzept. Es handelt sich um eine äußere Retinopathie, die trotz nahezu normalem Fundus zu plötzlichem Sehverlust, Gesichtsfeldausfällen und Photopsie führt3).
Der von Jampol et al. vorgeschlagene AZOOR-Komplex ist ein Konzept, das MEWDS, AZOOR, PIC, MFC, AMN (akute makuläre Neuroretinopathie), AIBSE und AAOR als ein Kontinuum mit gemeinsamer autoimmuner/entzündlicher genetischer Grundlage versteht3).
Charakteristische klinische Merkmale
Tritt vorwiegend bei jungen myopen Frauen im Alter von 20–50 Jahren auf.
Photopsie (Lichtsehen) tritt häufig im Frühstadium auf (insbesondere band- oder bogenförmige Lichter)
Beginnt einseitig und wird schließlich in 76 % der Fälle beidseitig
Der Augenhintergrund ist in der akuten Phase nahezu normal (charakteristisch ist die Diskrepanz zwischen Sehverschlechterung und Fundusbefund)
Gesichtsfeldausfälle zeigen ein unregelmäßiges bandförmiges Muster (oft an den blinden Fleck angrenzend)
Autoimmunerkrankungen (Hashimoto-Thyreoiditis, Multiple Sklerose) können begleitend auftreten
Das Verschwinden oder die Unschärfe der Ellipsoidzone (IS/OS-Linie) im OCT ist der wichtigste Befund.
Eine funktionelle Erholung der Bereiche, in denen die äußere Schicht im OCT verschwunden ist, ist nicht zu erwarten (auch für die Prognose nützlich).
Eine Amplitudenminderung im multifokalen ERG kann auch bei normalem Augenhintergrund nachgewiesen werden (multifokales ERG ist empfindlicher als Ganzfeld-ERG).
Infrarot-FAF kann manchmal die Grenze zwischen Läsion und normalem Bereich darstellen.
Behandlung und Prognose
Es gibt keine etablierte Behandlung für AZOOR. Leichte Fälle können nur beobachtet werden, aber schwere Fälle (Sehverschlechterung oder ausgedehnter Gesichtsfeldausfall) werden mit einer Methylprednisolon-Pulstherapie (1.000 mg × 3 Tage) gefolgt von oralem Prednisolon behandelt2). Die meisten Fälle stabilisieren sich innerhalb von 6 Monaten, aber das Gesichtsfeld erholt sich nicht in Bereichen mit verbleibenden Schäden der äußeren Schicht.
QWie unterscheidet man AZOOR von retrobulbärer Neuritis?
A
AZOOR und retrobulbäre Neuritis müssen unterschieden werden, da beide einen nahezu normalen Fundus mit verminderter Sehschärfe und Gesichtsfeldausfällen aufweisen. Die Unterscheidungspunkte sind: ① Bei AZOOR liegt eine verminderte Amplitude im multifokalen ERG vor, während bei retrobulbärer Neuritis das ERG normal ist; ② Bei AZOOR zeigen die Gesichtsfeldausfälle ein unregelmäßiges band- oder bogenförmiges Muster, während bei retrobulbärer Neuritis ein Zentralskotom häufig ist; ③ Bei AZOOR ist der RAPD in der Regel gering, während bei retrobulbärer Neuritis ein deutlicher RAPD vorliegt; ④ Im OCT zeigt AZOOR ein Verschwinden der Ellipsoidzone, während retrobulbäre Neuritis eine Papillenschwellung oder RNFL-Verdünnung zeigt, was zur Unterscheidung beiträgt2, 3).
Die klare Erfassung der Rolle jeder bildgebenden Untersuchung ist für die genaue Diagnose und Aktivitätsbeurteilung des White-Dot-Syndroms unerlässlich1, 5).
Beurteilung von Leckagen der Netzhautgefäße, des RPE und der Choriokapillaris. Bestätigung des Fluoreszenz-Umkehrphänomens bei APMPPE. Bestätigung der kranzförmigen Hyperfluoreszenz bei MEWDS. Beurteilung von Vaskulitis (Birdshot).
Direkte Beurteilung von Störungen der Aderhautdurchblutung. Frühere Erkennung von Läsionen als FA (insbesondere Birdshot, APMPPE, serpiginös). Erkennung von ausgedehnteren Läsionen als klinisch ersichtlich (MEWDS, PIC). Am empfindlichsten für die Erkennung aktiver Läsionen.
Nicht-invasive Beurteilung von RPE-Schäden. Aktivitätsbestimmung (APMPPE, Serpiginosa). Diagnose von MEWDS (frühe weiße Punkte mit hoher Autofluoreszenz). Beurteilung der Chronizität bei Birdshot (peripapilläre Hypoautofluoreszenz 73%)
Beurteilung der Ellipsoidzone (diagnostische Befunde bei MEWDS und AZOOR). 5-stufige Beurteilung der Läsionsevolution (PIC). Beurteilung von CNV und Makulaödem. Prognose (EZ-Verlust → schlechte Sehprognose)
OCT-A
Nicht-invasiver Nachweis von Choriokapillaris-Flow-Voids (APMPPE, Serpiginosa, PIC). Früher und sensitiver Nachweis von CNV (höhere Sensitivität als FA bei PIC und MFC). Schichtspezifische Beurteilung des choroidalen Blutflusses bei Birdshot. Therapieüberwachung
Diagnose von AZOOR (ERG-Amplitudenreduktion auch bei nahezu normalem Fundus). Aktivitätsüberwachung bei Birdshot (30-Hz-Flicker-Verzögerung am sensitivsten). Beurteilung des Therapieansprechens.
OCT-A (Suche nach Choriokapillaris-Flow-Void und CNV)
QSollte zuerst FA oder ICGA durchgeführt werden?
A
Die Strategie variiert je nach Erkrankung. Bei APMPPE und serpiginöser Choroiditis stellt die ICGA choroidale Durchblutungsstörungen direkter dar, daher vertieft die Durchführung der ICGA gleichzeitig oder nach der FA das Verständnis der Pathologie. Bei MEWDS ist die kranzförmige Hyperfluoreszenz in der FA diagnostisch wichtig. Da es sich jedoch um invasive Untersuchungen handelt, liefert die OCT-A mittlerweile viele alternative Informationen, und die Kombination von FAF und OCT-A wird als Erstbewertung genutzt1, 5).
Bedeutung von hypofluoreszenten Läsionen in der ICGA
Die Hypofluoreszenz von White-Dot-Syndrom-Läsionen in der ICGA ist ein direkter Hinweis auf eine Unterbrechung des choroidalen Blutflusses (Choriokapillaris-Verschluss)1). Die ICGA ist als Modalität zur Beurteilung des choroidalen Kreislaufs empfindlicher als die FA und weist folgende Merkmale auf:
Totale Hypofluoreszenz: tritt bei APMPPE, serpiginöser Choroiditis, PIC und MFC auf. Sie spiegelt einen schweren Verschluss der choroidalen Kapillaris-Ischämie wider.
Späte Hypofluoreszenz (FA normal) : Die späte Hypofluoreszenz im ICGA bei MEWDS wird durch eine veränderte ICG-Aufnahme aufgrund einer RPE-Anomalie erklärt, nicht durch eine Beteiligung der Choriokapillaris (die Choriokapillaris ist im OCT-A in der Regel erhalten)1).
Flow Void der Haller-Schicht → im fortgeschrittenen Stadium vollschichtiges Flow Void : Charakteristisches zweistufiges Fortschreiten bei Birdshot, beginnend im Aderhautstroma und dann auf die Choriokapillaris übergreifend14).
ICGA ist auch überlegen bei der Erkennung klinisch unsichtbarer subklinischer Läsionen im Vergleich zu FA und OCT-A, insbesondere bei MEWDS und PIC zeigt sie ausgedehntere Aderhautläsionen als die weißen Punkte1, 15).
Krankheitskarte nach Hypo- und Hyperfluoreszenzmustern in der FAF
Die Muster der FAF (Fundusautofluoreszenz) spiegeln den Stoffwechselzustand des RPE wider und sind nützlich für die Diagnose und Aktivitätsbeurteilung von White-Dot-Syndromen16).
Die Choriocapillaris-Flow-Void in der OCT-A zeigt eine hohe Übereinstimmung mit FA- und ICGA-Befunden (besonders nützlich bei APMPPE und serpiginöser Chorioiditis) 5, 13).
Fluoreszenzmuster in der FA: Füllungsverzögerung vs. Hyperfluoreszenz vs. Leckage
Starke Tendenz zur spontanen Besserung, oft ohne spezifische Behandlung2)
MEWDS: Weiße Punkte verschwinden innerhalb weniger Wochen, Sehkraft erholt sich. Nur bei schweren Fällen oder begleitendem Papillenödem orale Steroide.
APMPPE: Weiße Flecken bilden sich innerhalb von 7–12 Tagen zurück, Sehprognose in der Regel gut.
Erkrankungen, bei denen die Steroidtherapie im Vordergrund steht
Da die MFC einen chronisch-rezidivierenden Verlauf nimmt, ist nach Ausschleichen der Steroide häufig eine immunmodulatorische Erhaltungstherapie erforderlich.
QReicht eine Anti-VEGF-Therapie allein bei CNV aus?
A
Die entzündliche CNV (iCNVM) unterscheidet sich von der CNV bei altersbedingter Makuladegeneration; die Kontrolle der zugrunde liegenden Entzündung ist auch wichtig, um ein Wiederauftreten der CNV zu verhindern. Bei PIC und MFC wird ein bidirektionaler Ansatz mit Anti-VEGF und Steroiden (oder Immunsuppressiva) als wirksam angesehen; eine alleinige Anti-VEGF-Therapie birgt ein Rezidivrisiko 7, 8).
APMPPE (Kopfschmerzen, neurologische Symptome → notfallmäßige MRT des Gehirns)
QWelche Untersuchungen sollten zuerst durchgeführt werden, wenn Immunsuppressiva eingesetzt werden?
A
Vor Beginn einer Immunsuppression bei White-Dot-Syndrom sind folgende obligatorische Untersuchungen durchzuführen: ① Tuberkuloseausschluss mittels IGRA (Quantiferon) (höchste Priorität bei serpiginöser Chorioiditis), ② Virushepatitis-Screening mittels HBs-Antigen, HBc-Antikörper und HCV-Antikörper (Verhinderung einer Reaktivierung unter Immunsuppression), ③ Röntgen-Thorax und CT (Ausschluss von Tuberkulose und Sarkoidose), ④ großes Blutbild sowie Leber- und Nierenfunktionstests (Bestimmung der Ausgangswerte). Bei Biologika wie Adalimumab ist das Tuberkulose-Screening laut Packungsbeilage ebenfalls obligatorisch19, 20).
QWie werden Anzahl und Verabreichungsplan der Anti-VEGF-Therapie festgelegt?
A
Im Gegensatz zur altersbedingten Makuladegeneration kann eine entzündliche CNV (iCNVM) bei guter Entzündungskontrolle spontan zurückgehen. Üblicherweise wird eine PRN-Strategie (bei Bedarf) angewendet, wobei injiziert wird, sobald eine aktive CNV im OCT-A bestätigt wird. Eine initiale Aufsättigungsdosis (3 aufeinanderfolgende Injektionen) kann erfolgen, aber in Kombination mit einer Immunsuppression kann die Anzahl der benötigten Injektionen reduziert werden. Bei alleiniger Anti-VEGF-Therapie ist das Rezidivrisiko hoch, daher ist ein bidirektionaler Ansatz mit Kontrolle der zugrunde liegenden Entzündung wichtig 7, 8).
Jede Erkrankung des White-Dot-Syndroms ist mit bestimmten systemischen Erkrankungen oder Infektionen assoziiert, daher ist eine systematische Ausschlussdiagnostik vor der Behandlung wichtig.
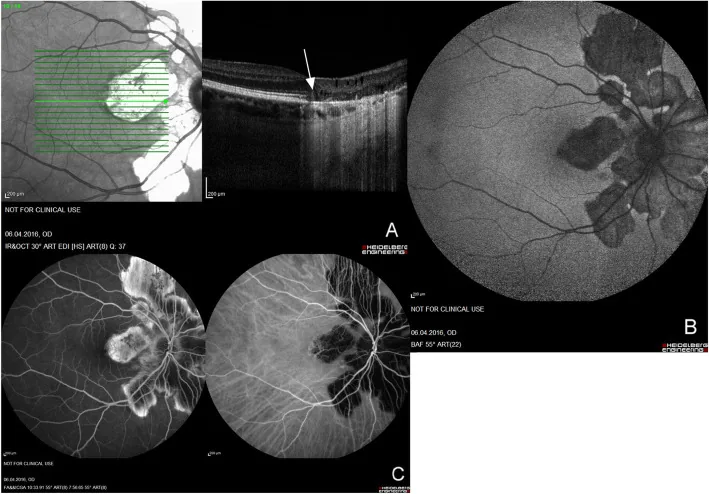
Die APMPPE wird heute zusammen mit der persistierenden Plakoid-Makulopathie (PPM) und der unerbittlichen Plakoid-Chorioretinitis (RPC) als „Plakoid-Chorioretinitis-Spektrum“ integriert verstanden. Bei diesen drei Erkrankungen zeigt die OCT-A ein gemeinsames Muster von Choriokapillaris-Flow-Void, was darauf hindeutet, dass eine Ischämie der Choriokapillaris eine gemeinsame pathologische Grundlage darstellt5).
Klufas et al. (2017) berichteten, dass OCT-A bei den drei Erkrankungen APMPPE, PPM und RPC mit hoher Übereinstimmung mit FA und ICGA Choriocapillaris-Flow-Voids nachweist und damit das Konzept des Placoid-Chorioretinitis-Spektrums unterstützt5).
Die foveale Granularität ist ein diagnostischer Befund, der bei 74–96 % der MEWDS-Patienten auftritt und nach dem Verschwinden der weißen Punkte als einziger Befund bestehen bleiben kann. Die Nahinfrarot-FAF (NIR-FAF) zeigt ein charakteristisches foveales granuläres Muster9).
PIC und MFC (MFCwP) teilen einen gemeinsamen genetischen Hintergrund (IL-10-Haplotyp, HLA-DRB1*15) und werden als unterschiedliche Phänotypen desselben Krankheitsspektrums angesehen. Die wichtigsten Unterscheidungsmerkmale sind das Vorhandensein oder Fehlen von Vitritis und Vorderkammerentzündung sowie die Verteilung der Läsionen1, 3).
Die Assoziation zwischen Birdshot und HLA-A29 ist eine der stärksten bekannten genetischen Assoziationen bei allen Erkrankungen, mit einem relativen Risiko von 50- bis 224-fach erhöht bei weißen Patienten4). Bei Japanern ist jedoch die Sensitivität der HLA-A29-Positivität für die Diagnose aufgrund der geringen Prävalenz von HLA-A29-Trägern begrenzt. Die Diagnose sollte sich auf die klinischen Befunde der SUN-2021-Klassifikationskriterien (Fundusbefunde, geringe Vorderkammerentzündung, Glaskörperentzündung) stützen10).
Es wird angenommen, dass AZOOR entsteht, wenn zu einer genetischen Prädisposition (IL-10-Haplotyp usw.) ein Umweltauslöser (Virusinfektion, Impfung, Medikament usw.) hinzukommt, und wird zusammen mit MEWDS, PIC, AMN und AIBSE als AZOOR-Komplex verstanden3). Weltweit nehmen Fälle von MEWDS nach COVID-19-Infektion oder Impfung zu, was darauf hindeutet, dass SARS-CoV-2 als Immunauslöser fungieren könnte11).
QKann das White-Dot-Syndrom nach einer COVID-19-Infektion oder Impfung auftreten?
A
Ja. Es wurden mehrere Fälle von Erstmanifestation oder Rezidiv von MEWDS, PIC und serpiginöser Chorioiditis nach einer COVID-19-Infektion berichtet, was darauf hindeutet, dass eine SARS-CoV-2-Infektion als immunologischer Auslöser fungieren könnte11, 12). Bei MEWDS gibt es eine systematische Übersicht über 27 Fälle nach COVID-19-Impfung, wobei der mRNA-Impfstoff (Pfizer-BioNTech) am häufigsten war. Personen mit entsprechender Vorgeschichte sollten vor und nach der Impfung einen Augenarzt zur Verlaufskontrolle konsultieren.
QWelche Untersuchungen sollten bei erstmaligem Verdacht auf ein White-Dot-Syndrom priorisiert durchgeführt werden?
A
Die empfohlene Priorität der Untersuchungen bei erstmaligem Verdacht auf ein White-Dot-Syndrom ist wie folgt: ① FAF + OCT (nicht-invasiv, ermöglicht die initiale Beurteilung fast aller Erkrankungen; Darstellung der Hyper-AF bei MEWDS, Beurteilung der Ellipsoidzone, sub-RPE-Erhebung bei PIC) → ② OCT-A (frühe Erkennung einer CNV, Beurteilung von choriokapillären Flow-Voids) → ③ FA + ICGA (falls eine definitive Diagnose oder Aktivitätsbeurteilung erforderlich ist). Bei Verdacht auf serpiginöse Chorioiditis sollte vor der FAIGRA (Tuberkulose-Ausschluss) priorisiert werden. Bei Verdacht auf AZOOR ist ein ERG (multifokales ERG) obligatorisch1, 2).
Testi I, Modugno RL, Pavesio C. Multimodal imaging supporting the pathophysiology of white dot syndromes. Journal of ophthalmic inflammation and infection. 2021;11(1):32. doi:10.1186/s12348-021-00261-3. PMID:34529201; PMCID:PMC8446150.
Jampol LM, Becker KG. White spot syndromes of the retina: a hypothesis based on the common genetic hypothesis of autoimmune/inflammatory disease. American journal of ophthalmology. 2003;135(3):376-9. doi:10.1016/s0002-9394(02)02088-3. PMID:12614757.
Agrawal R, et al. The role of HLA-A29 in birdshot chorioretinopathy and immune checkpoint inhibitor-related uveitis. Am J Ophthalmol. 2025. doi:10.1016/j.ajo.2024.01.007
Klufas MA, Phasukkijwatana N, Iafe NA, Prasad PS, Agarwal A, Gupta V, et al. Optical Coherence Tomography Angiography Reveals Choriocapillaris Flow Reduction in Placoid Chorioretinitis. Ophthalmology. Retina. 2017;1(1):77-91. doi:10.1016/j.oret.2016.08.008. PMID:31047399.
Stattin M, Forster J, Ahmed D, Krepler K, Ansari-Shahrezaei S. Swept Source-Optical Coherence Tomography Angiography for Management of Secondary Choroidal Neovascularization in Punctate Inner Choroidopathy. Case reports in ophthalmology. 2021;12(1):232-238. doi:10.1159/000511669. PMID:33976688; PMCID:PMC8077444.
Spaide RF, Goldberg N, Freund KB. Redefining multifocal choroiditis and panuveitis and punctate inner choroidopathy through multimodal imaging. Retina (Philadelphia, Pa.). 2013;33(7):1315-24. doi:10.1097/IAE.0b013e318286cc77. PMID:23584703.
Leclaire MD, Clemens CR, Eter N, Mihailovic N. Choroidale Neovaskularisation infolge einer “punctate inner choroidopathy”, dargestellt mittels optischer Kohärenztomographie-Angiographie. Ophthalmologe. 2021;118:842-846. doi:10.1007/s00347-020-01200-8.
Mantovani A, Invernizzi A, Staurenghi G, Herbort CP.. Multiple Evanescent White Dot Syndrome: A Multimodal Imaging Study of Foveal Granularity. Ocul Immunol Inflamm. 2019;27(1):141-147. doi:10.1080/09273948.2017.1353104. PMID:28981397.
Standardization of Uveitis Nomenclature (SUN) Working Group. Classification Criteria for Birdshot Chorioretinitis. American journal of ophthalmology. 2021;228:65-71. doi:10.1016/j.ajo.2021.03.059. PMID:33845003; PMCID:PMC8517033.
Chen N, Mandell M, Arjmand P. Multimodal imaging findings of multiple evanescent white dot syndrome in COVID-19 patients. IDCases. 2024;38:e02110. doi:10.1016/j.idcr.2024.e02110. PMID:39582747; PMCID:PMC11585666.
Seddigh S, Pinto A, Zaki AM, Gupta RR. Serpiginous Choroiditis After COVID-19 Infection. Journal of vitreoretinal diseases. 2025;9(2):246-252. doi:10.1177/24741264241297936. PMID:39554631; PMCID:PMC11562243.
Pakzad-Vaezi K, Khaksari K, Chu Z, Van Gelder RN, Wang RK, Pepple KL.. Swept-Source OCT Angiography of Serpiginous Choroiditis. Ophthalmol Retina. 2018;2(7):712-719. doi:10.1016/j.oret.2017.11.001. PMID:30148243; PMCID:PMC6103638.
Pepple KL, Chu Z, Weinstein J, Munk MR, Van Gelder RN, Wang RK. Use of en face swept-source optical coherence tomography angiography in identifying choroidal flow voids in 3 patients with birdshot chorioretinopathy. JAMA Ophthalmol. 2018;136(11):1288-1292. doi:10.1001/jamaophthalmol.2018.3474. PMID:30128478. PMCID:PMC6248174
Khochtali S, Dridi T, Abroug N, Ksiaa I, Lupidi M, Khairallah M. Swept-Source Optical Coherence Tomography Angiography Shows Choriocapillaris Flow Reduction in Multiple Evanescent White Dot Syndrome. Journal of current ophthalmology. 2020;32(2):211-215. doi:10.4103/JOCO.JOCO_107_20. PMID:32671309; PMCID:PMC7337020.
Yeh S, Forooghian F, Wong WT, Faia LJ, Cukras C, Lew JC, Wroblewski K, Weichel ED, Meyerle CB, Sen HN, Chew EY, Nussenblatt RB.. Fundus autofluorescence imaging of the white dot syndromes. Arch Ophthalmol. 2010;128(1):46-56. doi:10.1001/archophthalmol.2009.368. PMID:20065216; PMCID:PMC3025103.
Minos E, Barry RJ, Southworth S, et al. Birdshot chorioretinopathy: current knowledge and new concepts in pathophysiology, diagnosis, monitoring and treatment. Orphanet J Rare Dis. 2016;11:61. doi:10.1186/s13023-016-0429-8. PMID:27175923; PMCID:PMC4866419.
Maleki A, Maldonado Cerda A, Garcia CM, et al. Chlorambucil combination therapy in refractory serpiginous choroiditis: a cure? Am J Ophthalmol Case Rep. 2021;21:101014. doi:10.1016/j.ajoc.2021.101014. PMID:33615036; PMCID:PMC7881218.
Niederer RL, Al-Janabi A, Engelbrecht C, et al. Immunomodulatory therapy prescribing practices for non-infectious uveitis: a survey of international experts. Br J Ophthalmol. 2024;108:482-489.
Tomkins-Netzer O, et al. Treatment of non-infectious uveitis with biologics: a survey of the International Ocular Inflammation Society. Br J Ophthalmol. 2022;106:482-488.
Kopieren Sie den Artikeltext und fügen Sie ihn in den KI-Assistenten Ihrer Wahl ein.
Artikel in die Zwischenablage kopiert
Öffnen Sie unten einen KI-Assistenten und fügen Sie den kopierten Text in den Chat ein.