Die punktförmige innere Choroidopathie (punctate inner choroidopathy) ist eine idiopathische entzündliche Aderhauterkrankung, die erstmals 1984 von Watzke et al. beschrieben wurde. Sie wird zu den White-Dot-Syndromen gezählt und gehört zu den entzündlichen Erkrankungen, die die äußere Netzhaut, die Choriokapillaris und die Aderhaut betreffen 7). In den Leitlinien zur Uveitis-Diagnostik und -Therapie (Jpn J Ophthalmol 2019;123(6):635-696) wird sie als nicht-infektiöse, hintere Uveitis mit Fundusbeteiligung eingeordnet, mit den Merkmalen eines schleichenden Beginns und ein- oder beidseitigem Auftreten 8).
Sie tritt hauptsächlich bei jungen Frauen mit Kurzsichtigkeit auf (etwa 90 % sind weiblich) 5). Große Studien berichten ein durchschnittliches Erkrankungsalter von 36 Jahren und eine mittlere Myopie von −4,5 Dioptrien 5). Sie bevorzugt junge Frauen mit mittlerer Myopie; im akuten Stadium finden sich mehrere kleine gelb-weiße Herde im hinteren Pol, die im Laufe der Zeit zu atrophischen Läsionen werden.
Punktförmige innere Choroidopathie und multifokale Choroiditis (MFC) haben gemeinsam, dass sie die Aderhaut, das retinale Pigmentepithel (RPE) und die äußere Netzhaut betreffen, was auf ein mögliches gemeinsames Krankheitsspektrum hindeutet 7). Die Unterscheidung zwischen beiden Erkrankungen erfolgt anhand des Vorhandenseins einer Glaskörperentzündung und der Verteilung der Läsionen. Die punktförmige innere Choroidopathie geht ohne Glaskörperentzündung einher, und die Läsionen sind auf den hinteren Pol beschränkt, während die MFC mit einer Glaskörperentzündung einhergeht und die Läsionen auch die Peripherie betreffen 6).
QWas ist der Unterschied zwischen punktförmiger innerer Choroidopathie und multifokaler Choroiditis (MFC)?
A
Die punktförmige innere Choroidopathie geht ohne Glaskörperentzündung einher, und die Läsionen sind auf den hinteren Pol beschränkt, während die MFC mit einer Glaskörperentzündung und Vorderkammerentzündung einhergeht und die Läsionen auch die mittlere Peripherie betreffen. Es wird auch darauf hingewiesen, dass beide Erkrankungen möglicherweise ein gemeinsames Spektrum darstellen.
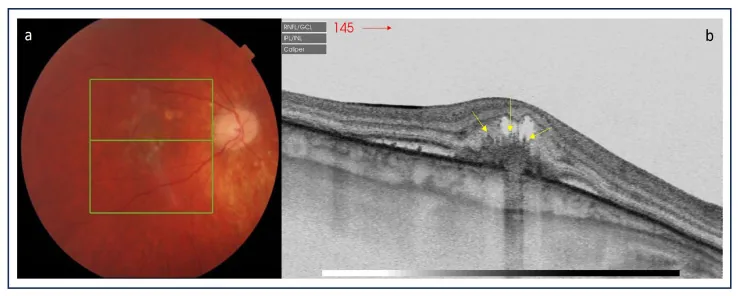
Karska-Basta I, et al. Diagnostic Challenges in Inflammatory Choroidal Neovascularization. Medicina (Kaunas). 2024. Figure 5. PMCID: PMC10972505. License: CC BY.
Im linken Fundusfoto sind gelb-weiße kleine Läsionen im hinteren Pol zu sehen. Im rechten OCT zeigen sich hyperreflektive Läsionen in der Makula sowie subretinale und intraretinale Flüssigkeit, was auf aktive PIC-Läsionen hinweist.
Das häufigste Erstsymptom der punktförmigen inneren Choroidopathie sind einseitige Skotome und Sehverschlechterung.
Verschlechterung der Sehschärfe: Die Sehschärfe bei der Erstuntersuchung reicht von 0,4 bis 0,05. Berichten zufolge behielten etwa 67 % eine korrigierte Sehschärfe von 0,4 oder besser. In der Remissionsphase verbleiben scharf begrenzte runde atrophische Herde mit Pigmentierung 8).
Skotome: Es werden zentrale oder parazentrale Skotome wahrgenommen.
Photopsien: Es können Lichtblitze auftreten.
Metamorphopsien: Es kann über verzerrtes Sehen geklagt werden.
Mouches volantes: Bei einigen Patienten vorhanden.
Klinische Befunde (vom Arzt bei der Untersuchung festgestellte Befunde)
Als Fundusbefund finden sich 12 bis 25 kleine gelb-weiße Flecken von 100–300 μm Größe, die auf den hinteren Pol beschränkt sind und in einem zufälligen Muster verteilt sind. Die Läsionen treten auf der Ebene der äußeren Netzhaut, des RPE und der inneren Aderhaut auf. 80 % sind bilateral, aber oft asymmetrisch 1).
Die Merkmale der punktförmigen inneren Aderhauterkrankung sind im Folgenden zusammengefasst.
Aktive Läsionen
Gelblich-weiße chorioretinale Läsionen: Am hinteren Pol finden sich kleine, scharf begrenzte gelbgraue Flecken. Die Papillenregion bleibt ausgespart.
Fehlen einer intraokularen Entzündung: Das Fehlen von Vorderkammerentzündung und Glaskörperentzündung ist charakteristisch für die punktförmige innere Aderhautentzündung 3).
Neurosenkung der Netzhautablösung: Über aktiven Läsionen kann eine seröse Netzhautablösung auftreten.
Narbenstadium-Läsion
Atrophische chorioretinale Narbe: Nach Abklingen der Entzündung hinterlässt sie eine scharf begrenzte „ausgestanzte“ Narbe. Begleitet von Pigmentierung und depigmentiertem Halo.
Narbenausdehnung im Laufe der Zeit: Auch nach Abklingen der Entzündung dehnt sich die Narbe allmählich aus und kann in der Nähe der Fovea zu einer Verschlechterung der Sehkraft führen.
Chorioidale Neovaskularisation (CNV): Tritt bei 40–76 % der Fälle auf5) und ist die Hauptursache für Sehverschlechterung.
Die SD-OCT zeigt eine fünfstufige Evolution der punktierten inneren Aderhauterkrankung 2).
Die Ätiologie der punktierten inneren Aderhauterkrankung ist unbekannt, aber es wird angenommen, dass es sich um eine Autoimmunerkrankung mit polygenetischer Anfälligkeit handelt, die durch Umweltreize wie Infektionen, Impfungen oder Stress ausgelöst wird.
Es wurde über das Auftreten und die Reaktivierung einer punktierten inneren Aderhauterkrankung nach einer COVID-19-Infektion berichtet. Eine SARS-CoV-2-Infektion kann bei Personen mit genetischer Veranlagung eine Fehlregulation des Immunsystems auslösen. Darüber hinaus wurde auch ein Wiederaufflammen der punktierten inneren Aderhauterkrankung nach einer COVID-19-Impfung beschrieben3).
Scott et al. (2024) berichteten über eine 38-jährige Frau mit einem Rezidiv einer punktierten inneren Aderhauterkrankung und einer entzündlichen choroidalen Neovaskularisationsmembran (iCNVM) 7 Tage nach der Pfizer-BioNTech COVID-19-Impfung. Unter Immunsuppression trat kein Rezidiv auf, jedoch nach der vierten Impfung ohne Immunsuppression 3).
QKann die COVID-19-Impfung eine punktierte innere Aderhauterkrankung verschlimmern?
A
Nach der COVID-19-Impfung wurden Rezidive einer punktierten inneren Aderhauterkrankung berichtet 3). Der Nutzen der Impfung überwiegt jedoch das Risiko eines Augenentzündungsrezidivs bei weitem. Betroffene mit Vorgeschichte sollten vor und nach der Impfung die Nachsorge mit einem Augenarzt besprechen.
Die Diagnose der punktförmigen inneren Choroidopathie basiert auf klinischen Befunden. Die definitive Diagnose wird durch ergänzende bildgebende Untersuchungen gestellt. Der Histoplasmin-Hauttest ist negativ.
In der FA zeigen aktive Läsionen in der frühen arteriellen Phase eine Hyperfluoreszenz und in der späten Phase Leckage und Anfärbung 7). Es werden mehr Läsionen nachgewiesen als bei der klinischen Untersuchung. Die CNVM stellt sich als unregelmäßiges, netzartiges Gefäßnetzwerk dar.
In der ICG zeigen sich mittlere hypofluoreszente Läsionen im hinteren Pol und peripapillär 7). Bei 32% der betroffenen Augen werden subklinische hypofluoreszente Flecken nachgewiesen, was die diagnostische Genauigkeit erhöht.
Die SD-OCT ist für die Diagnose und Verlaufskontrolle der punktförmigen inneren Choroidopathie nützlich. Bei aktiven Läsionen zeigen sich lokale hyperreflektive Erhebungen des RPE und eine Unterbrechung der Ellipsoidzone (EZ) 7). Choroidale Neovaskularisations-positive Läsionen haben im Vergleich zu choroidalen Neovaskularisations-negativen Läsionen eine größere Höhe, Breite und Volumen, begleitet von einer Unterbrechung der EZ und Bruch-Membran sowie einer Unschärfe der äußeren Netzhaut.
In der tiefen Bildgebung OCT (EDI-OCT) zeigt sich eine erhöhte Aderhautdicke direkt unter aktiven Läsionen, die nach der Behandlung abnimmt („Schwammzeichen“). Dies ermöglicht die Unterscheidung von myopischer choroidaler Neovaskularisation.
Die OCTA ist eine wichtige Bildgebungsmodalität bei der Diagnose der punktierten inneren Aderhauterkrankung. Sie kann sekundäre choroidale Neovaskularisationen, die mit herkömmlicher FA oder SD-OCT schwer nachweisbar sind, mit hoher Rate identifizieren.
Mit der OCTA kann bei vielen Patienten mit punktierter innerer Aderhauterkrankung, einschließlich solcher, bei denen die FA keine eindeutigen Ergebnisse lieferte, das Vorhandensein einer choroidalen Neovaskularisation nachgewiesen werden.
Leclaire et al. (2021) berichteten über Fälle, bei denen sekundäre choroidale Neovaskularisationen, die in der FAG/SD-OCT nicht nachweisbar waren, nur mittels OCT-A identifiziert wurden, was auf eine mögliche hohe Dunkelziffer klinisch asymptomatischer sekundärer choroidaler Neovaskularisationen hindeutet5).
Stattin et al. (2021) berichteten, dass sie mittels SS-OCTA die Veränderungen der Gefäßdichte bei choroidalen Neovaskularisationen infolge einer punktierten inneren Aderhauterkrankung überwachten und dies als Entscheidungsgrundlage für die Anti-VEGF-Therapie nutzten4).
Aktive Läsionen der punktierten inneren Aderhauterkrankung stellen sich als hypoautofluoreszierend dar1). Ein hyperautofluoreszierender Hof um die aktive Läsion könnte ein indirektes Zeichen für eine unkontrollierte Entzündung sein. Atrophische Narbenläsionen sind ebenfalls als hypoautofluoreszierende Flecken erkennbar1).
Die Fundus-Autofluoreszenz ist eine nicht-invasive, schnelle Untersuchung, die für die Beurteilung der Läsionsverteilung, die Überwachung des Therapieerfolgs und die Erkennung von Rezidiven nützlich ist1).
Etwa 41 % zeigen eine Vergrößerung des blinden Flecks, auch zentrale und parazentrale Skotome werden beobachtet. 45 % der Patienten weisen ein normales Gesichtsfeld auf.
Die folgenden Erkrankungen sollten differenzialdiagnostisch in Betracht gezogen werden:
Multiple evanescent white dot syndrome (MEWDS): Meist einseitig und spontan rückbildungsfähig. Charakteristisch ist eine kranzförmige Hyperfluoreszenz6).
Subretinale Fibrose und Uveitis-Syndrom: Geht mit progressiver subretinaler Fibrose einher 8).
Okuläre Toxoplasmose / okuläre Tuberkulose: Als infektiöse Erkrankungen müssen sie ausgeschlossen werden 8).
Fuchs-Fleck bei hoher Myopie: Als degenerative Erkrankung differenzialdiagnostisch abzugrenzen 8).
QWas ist der Unterschied zwischen punktförmiger innerer Choroidopathie und multiplen evaneszenten weißen Punkten?
A
Das Syndrom der multiplen evaneszenten weißen Punkte ist meist einseitig, bildet sich innerhalb weniger Wochen spontan zurück und hinterlässt kaum Narben oder choroidale Neovaskularisation6). Die punktförmige innere Choroidopathie ist häufig beidseitig, hinterlässt atrophische Narben und ist häufig mit choroidaler Neovaskularisation assoziiert. Auch das Vorhandensein von Blutfluss in der OCTA kann zur Differenzierung beitragen.
Liegen keine Hinweise auf eine choroidale Neovaskularisation vor, ist die Sehprognose gut und eine Behandlung ist in den meisten Fällen nicht erforderlich. Die einzige Ausnahme sind aktive entzündliche Läsionen in unmittelbarer Nähe des Fixationspunktes, bei denen eine medikamentöse Behandlung in Betracht gezogen wird.
Bei aktiven Läsionen in der Nähe der Fovea wird eine subtenonale Injektion von Triamcinolonacetonid im hinteren Bereich oder eine orale Steroidtherapie (Prednisolon 40–60 mg/Tag mit anschließender Dosisreduktion) durchgeführt 8). Spricht eine extrafoveale choroidale Neovaskularisation nicht auf Steroide an, kann eine Photokoagulation versucht werden.
Dies ist die zentrale Behandlung der punktierten inneren Choroidopathie mit choroidaler Neovaskularisation. Bevacizumab, Ranibizumab und Aflibercept werden eingesetzt.
Stattin et al. (2021) verabreichten Ranibizumab (0,5 mg) pro re nata unter SS-OCTA-Überwachung bei choroidaler Neovaskularisation infolge einer punktierten inneren Choroidopathie und erreichten nach insgesamt 6 Injektionen einen endgültigen Visus von 20/20 4).
Ein bidirektionaler Ansatz mit Anti-VEGF-Medikamenten und Steroiden gilt als wirksam 4). Bei CNV-Komplikationen wird manchmal die intravitreale Injektion von Bevacizumab als erste Wahl eingesetzt 8).
Systemische Steroide werden in der Regel mit 1 mg/kg/Tag (60–80 mg/Tag) für 3–5 Tage begonnen und dann schrittweise reduziert 4). Es wird berichtet, dass die Anwendung oraler Steroide das Risiko einer iCNVM halbiert 3).
Zu den intravitrealen Steroidpräparaten gehören:
Intravitreale Triamcinolon-Injektion (4 mg): In Kombination mit photodynamischer Therapie (PDT) verbessert sich der logMAR BCVA von 0,52 auf 0,20.
Intravitreales Dexamethason-Implantat (0,7 mg/0,35 mg): Freisetzung über 6 Monate. Wird in Kombination mit Anti-VEGF eingesetzt.
Fluocinolonacetonid-Implantat (0,59 mg): Freisetzung über 36 Monate. Nützlich bei Unverträglichkeit systemischer Therapie oder bei Frauen mit Kinderwunsch.
Bei Patienten mit choroidaler Neovaskularisation wurde die Wirksamkeit der PDT berichtet. In Kombination mit oralem Prednisolon (1 mg/kg/Tag) wurde nach durchschnittlich zwei PDT-Sitzungen eine Verbesserung der Sehschärfe um 15 Buchstaben erzielt.
QWie viele Anti-VEGF-Injektionen sind bei choroidaler Neovaskularisation bei punktförmiger innerer Choroidopathie erforderlich?
A
Dies variiert je nach Fall. Unter SS-OCTA-gesteuerter Pro-re-nata-Gabe wurden sechs Injektionen berichtet 4). Regelmäßige Bildgebung zur Beurteilung der Aktivität der choroidalen Neovaskularisation ist erforderlich, und bei einem Rezidiv können zusätzliche Injektionen notwendig sein.
6. Pathophysiologie und detaillierter Krankheitsmechanismus
Die Pathophysiologie der punktierten inneren Aderhauterkrankung ist nicht vollständig geklärt. Die führende Hypothese ist, dass es sich um eine entzündliche Erkrankung handelt, die in der inneren Aderhaut ihren Ursprung nimmt.
Pathologische Studien der choroidalen Neovaskularisation (CNV) als Folge der punktierten inneren Aderhauterkrankung zeigen, dass die Choriokapillaris erhalten bleibt, während auf der Ebene der inneren Aderhaut eine lymphozytäre Infiltration vorliegt. Dieser Befund unterstützt ultrastrukturell die Hypothese, dass die punktierte innere Aderhauterkrankung eine entzündliche Erkrankung mit Ursprung in der Aderhaut ist.
In der ICG-Angiographie entsprechen hypofluoreszente Areale einer lokalen choroidalen Minderperfusion, und lokale hyperfluoreszente Punkte an den Gefäßwänden könnten auf eine Vaskulitis hindeuten. Da große Aderhautgefäße diese hypofluoreszenten Areale kreuzen, wird angenommen, dass der Vaskulitisprozess auf kleine Gefäße und die Choriokapillaris beschränkt ist.
Die mit der punktierten inneren Aderhauterkrankung assoziierte choroidale Neovaskularisation ist vom Typ 2 (über dem retinalen Pigmentepithel) und entsteht durch Schädigung der Bruch-Membran und des retinalen Pigmentepithels. Neovaskuläre Einheiten mit wenigen Perizyten zeigen eine hohe Empfindlichkeit gegenüber Anti-VEGF-Medikamenten, was Perizyten zu einem wichtigen therapeutischen Ziel machen könnte.
In der OCT-Angiographie zeigen aktive entzündliche chorioretinale Läsionen auf der Ebene der Choriokapillaris einen fehlenden Blutflusssignal-Nachweis 7). Ob eine primäre Beteiligung der Choriokapillaris den äußeren Netzhautveränderungen vorausgeht oder umgekehrt, ist weiterhin Gegenstand der Diskussion 7).
Bei Patienten mit White-Dot-Syndrom und deren Familien ist die Prävalenz systemischer Autoimmunerkrankungen hoch6). Bei Patienten mit punktförmiger innerer Aderhauterkrankung wurde das Vorhandensein von IL-10-Haplotypen und HLA-DRB1*15-Allelen berichtet, und es wird angenommen, dass die Erkrankung durch das Zusammenwirken genetischer Veranlagung und Umweltfaktoren ausgelöst wird6).
Jampol und Becker (2003) schlugen vor, MEWDS, MCP, PIC und AZOOR als „AZOOR-Komplex“ zu einem einzigen klinischen Konzept zusammenzufassen. Die Hypothese besagt, dass genetische Prädisposition in Kombination mit verschiedenen Umweltauslösern zu unterschiedlichen klinischen Phänotypen führt6).
Liu et al. (2024) berichteten über einen 91-monatigen Follow-up-Fall einer solitären punktierten Chorioretinitis (SPC), einer Subtyp der punktierten inneren Choroidopathie. SPC ist ein Subtyp, bei dem eine einzelne Läsion in der Nähe der Fovea centralis auftritt, und hat im Vergleich zur punktierten inneren Choroidopathie eine geringere sekundäre Inzidenz von choroidalen Neovaskularisationen (16 % vs. etwa 50 %). Während des 91-monatigen Verlaufs blieb die Läsion solitär, und die gefäßähnlichen Strukturen im OCTA bildeten sich spontan zurück. Die endgültige Sehschärfe erholte sich auf 0,8, und eine Anti-VEGF-Therapie war nicht erforderlich 2).
Stattin et al. (2021) zeigten mittels SS-OCTA-En-face-Bildern, dass Veränderungen der Gefäßstruktur (Verzweigungen, Schleifen, Anastomosen) choroidaler Neovaskularisationen im zeitlichen Verlauf verfolgt werden können und dass SS-OCTA Veränderungen der Neovaskularisationen erfassen kann, selbst wenn indirekte Aktivitätszeichen im SD-OCT fehlen 4). Die Beurteilung der choroidalen Neovaskularisationsaktivität und die Therapieentscheidung auf Basis von OCTA-Befunden könnten zukünftig zum Standardmanagement der punktförmigen inneren Choroidopathie werden.
Impfstoffbedingter Rückfall und prophylaktische Immunsuppression
Scott et al. (2024) berichteten, dass unter Immunsuppression bei COVID-19-Impfung kein Rückfall der punktförmigen inneren Choroidopathie auftrat 3). Die Rolle der prophylaktischen Immunsuppression bei der Impfung von Hochrisikopatienten ist Gegenstand zukünftiger Untersuchungen.
Koexistenz von punktförmiger innerer Choroidopathie und multiplem evaneszentem weißen Punktsyndrom
Walters et al. (2021) berichteten über einen seltenen Fall eines Patienten mit langjähriger punktförmiger innerer Choroidopathie, bei dem akut ein multiples evaneszentes weißes Punktsyndrom auftrat. Die Koexistenz von punktförmiger innerer Choroidopathie und multiplem evaneszentem weißen Punktsyndrom unterstützt das Konzept des AZOOR-Komplexes und deutet auf eine gemeinsame genetische Grundlage hin 6).
Olazaran L, Jiménez A, González de Los Mártires P, Guerrero G, Gangoitia N, Salmeron I, Galarza A, Argüelles AS, Elso B, Reyzabal I, Compains E, Heras H, López S.. White Dot Syndromes: Report of Three Cases. Case Rep Ophthalmol. 2024;15(1):202-211. doi:10.1159/000536336. PMID:38487796; PMCID:PMC10939511.
Liu C, Liu M, Lan X, Zhu J, Zhang Z.. 91-month follow-up of solitary punctate chorioretinitis in a Chinese patient. BMC Ophthalmol. 2024;24(1):297. doi:10.1186/s12886-024-03576-6. PMID:39030539; PMCID:PMC11264762.
Daniel Andrew Richard Scott, Rachael Louise Niederer. Punctate Inner Choroidopathy (PIC) disease recurrence with inflammatory choroidal neovascular membrane (iCNVM) post-COVID-19 vaccine. European Journal of Ophthalmology. 2024;34(5):NP78-NP82. doi:10.1177/11206721241257969.
Stattin M, Forster J, Ahmed D, Krepler K, Ansari-Shahrezaei S. Swept Source-Optical Coherence Tomography Angiography for Management of Secondary Choroidal Neovascularization in Punctate Inner Choroidopathy. Case reports in ophthalmology. 2021;12(1):232-238. doi:10.1159/000511669. PMID:33976688; PMCID:PMC8077444.
Leclaire MD, Clemens CR, Eter N, Mihailovic N. Choroidale Neovaskularisation infolge einer “punctate inner choroidopathy”, dargestellt mittels optischer Kohärenztomographie-Angiographie. Ophthalmologe. 2021;118:842-846. doi:10.1007/s00347-020-01200-8.
Walters AR, Choi RY, Flaxel CJ.. Multiple Evanescent White Dot Syndrome Presenting in a Patient With Punctate Inner Choroidopathy. J Vitreoretin Dis. 2021;5(3):270-274. doi:10.1177/2474126420965031. PMID:37006511; PMCID:PMC9979046.
Testi I, Modugno RL, Pavesio C. Multimodal imaging supporting the pathophysiology of white dot syndromes. Journal of ophthalmic inflammation and infection. 2021;11(1):32. doi:10.1186/s12348-021-00261-3. PMID:34529201; PMCID:PMC8446150.