As facomatoses (phakomatoses) são um termo geral para um grupo de doenças congênitas caracterizadas por lesões hamartomatosas na pele, sistema nervoso central e olhos. Também são chamadas de síndromes neurocutâneas (neurocutaneous syndromes).
A denominação foi dada pelo oftalmologista holandês Van der Hoeve, derivada do grego «phakos» (lente/mancha). Inicialmente incluía três doenças: neurofibromatose, esclerose tuberosa e doença de von Hippel-Lindau, posteriormente adicionadas a síndrome de Sturge-Weber e a ataxia telangiectásica. Atualmente, mais de 60 síndromes foram descritas.
A base fisiopatológica comum é a anormalidade na formação, migração e diferenciação das células da crista neural. As células da crista neural derivam do ectoderma e produzem diversas células, como células de Schwann e melanócitos, resultando em lesões em múltiplos órgãos, incluindo nervos, pele e olhos. As vias de sinalização envolvidas incluem RAS, MAPK/MEK, mTOR, PI3K/AKT, GNAQ e VHL-HIF.
As frequências de ocorrência das seis principais doenças são as seguintes:
As seis principais doenças são NF1, NF2, esclerose tuberosa, síndrome de Sturge-Weber, doença de von Hippel-Lindau e ataxia telangiectasia. Todas são causadas por mutações genéticas e produzem lesões nos sistemas nervoso, cutâneo e ocular. Atualmente, mais de 60 síndromes estão incluídas na categoria de facomatoses.
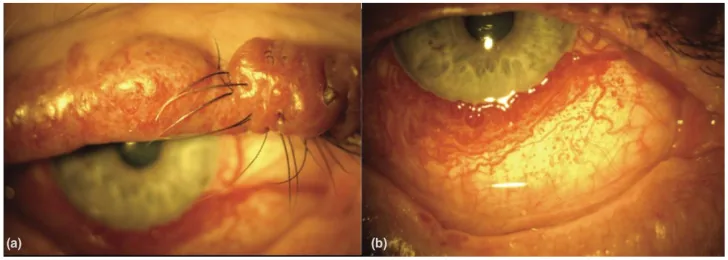
An Update on Multimodal Ophthalmological Imaging of Diffuse Choroidal Hemangioma in Sturge–Weber Syndrome. Vision (Basel). 2023 Oct 6; 7(4):64. Figure 1. PMCID: PMC10594527. License: CC BY.
(a) Mancha vinho do Porto na pálpebra superior com nodularidade em paciente com síndrome de Sturge–Weber. (b) Vascularização conjuntival difusa em paciente com síndrome de Sturge–Weber. De [15].
Os tipos de complicações oculares diferem em cada doença, e os sintomas subjetivos também são variados.
NF1: Com a progressão do tumor da via óptica (glioma óptico), ocorre diminuição da acuidade visual, perda da visão de cores e defeitos de campo visual. O neurofibroma plexiforme pode causar proptose.
Esclerose tuberosa: O hamartoma astrocítico retiniano geralmente é assintomático. Causa deficiência visual quando afeta a mácula ou o disco óptico.
SWS: Quando acompanhada de glaucoma congênito, ocorre opacidade corneana, lacrimejamento e fotofobia. No glaucoma de início tardio, ocorre defeito de campo visual progressivo indolor.
VHL: Assintomático no início. Com a progressão do hemangioblastoma capilar retiniano, surgem alterações exsudativas, edema macular e anel branco, levando à diminuição da acuidade visual.
AT: A visão geralmente é preservada. Os principais distúrbios são distúrbios do movimento ocular (nistagmo, apraxia oculomotora).
Nódulos de Lisch: Achado ocular mais frequente na NF1. Pequenos nódulos (1-2 mm) acastanhados, bem delimitados, em forma de cúpula, múltiplos na íris. Prevalência por idade: <5% em <3 anos, 42% em 3-4 anos, 55% em 5-6 anos, 100% em >21 anos. Incluído nos critérios diagnósticos de NF1 (≥2 nódulos). Como a íris dos japoneses é acastanhada, o exame com lâmpada de fenda é importante.
Tumores da via óptica (gliomas ópticos): Ocorrem em cerca de 15-25% dos pacientes com NF1. Frequentemente astrocitomas pilocíticos de baixo grau, geralmente assintomáticos. Quando progressivos, causam atrofia do nervo óptico, deficiência visual e defeitos de campo visual. Podem invadir o quiasma óptico.
Neurofibroma plexiforme: Ocorre em menos de 10% dos pacientes com NF1. Caracteriza-se por deformidade palpebral em forma de S e, à palpação, sensação de saco de vermes. Pode causar proptose, estrabismo, ambliopia e glaucoma congênito.
Displasia da asa maior do esfenoide: Defeito congênito da parede orbitária que pode causar proptose pulsátil.
Glaucoma: Ocorre em 1-2% dos pacientes com NF1. Existem dois tipos: congênito (unilateral) e de início tardio.
Outros: Proeminência dos nervos corneanos mielinizados, espessamento coroidal e fibras nervosas mielinizadas da retina.
Ceratite de exposição: Causada por disfunção dos nervos cranianos V e VII devido a neuroma acústico bilateral, resultando em dormência facial, diplopia e lagoftalmia.
Hamartoma astrocítico retiniano: Presente em cerca de 50% dos casos. Classificado em 3 tipos: (1) plano, translúcido, não calcificado, (2) elevado, multinodular, calcificado (aspecto de amora), (3) tipo transicional. Geralmente múltiplos no polo posterior.
A tríade principal é (1) hemangioma facial na área do nervo trigêmeo, (2) hemangioma intracraniano ipsilateral, (3) glaucoma ou hemangioma coroidal ipsilateral.
Glaucoma: O achado ocular mais importante na SWS. Ocorre em 30-70% dos casos. O glaucoma congênito (nascimento aos 4 anos) representa cerca de 60%, causando buftalmia, opacidade corneana e megalocórnea. A etiologia envolve disgenesia angular, aumento da pressão venosa epiescleral e hemangioma coroidal. Ocorre frequentemente na presença de hemangioma palpebral.
Hemangioma coroideu: Ocorre em cerca de 20-70% dos casos. Difícil de identificar no exame de fundo de olho comum por ser difuso e de limites imprecisos. O fundo de olho apresenta aspecto de molho de tomate. Geralmente não tende a crescer, mas pode causar alterações exsudativas ou descolamento exsudativo da retina.
Outros: Dilatação e tortuosidade dos vasos conjuntivais, episclerais e da íris.
Achados oculares na VHL (Doença de Von Hippel-Lindau)
Hemangioblastoma capilar da retina (angioma): Ocorre em 43-85% dos pacientes com VHL (cerca de 60% em relatos nacionais). Cerca de um terço é bilateral e múltiplo. Comumente localizado na região temporal periférica da retina como um nódulo alaranjado-avermelhado com vasos aferentes e eferentes dilatados e tortuosos. A idade média de início é 25 anos, geralmente aparecendo antes dos 30 anos.
Com a progressão, ocorrem alterações exsudativas, hemorragia retiniana → edema macular, anel branco → diminuição da acuidade visual.
Na angiografia fluoresceínica, observa-se fluxo do corante para a artéria aferente → veia eferente, com extravasamento precoce e acentuado do corante na massa tumoral.
Telangiectasia conjuntival bulbar: O achado ocular mais comum. Geralmente surge entre 5 e 8 anos de idade, presente em 80–90% dos pacientes1).
Distúrbios dos movimentos oculares: Nistagmo, apraxia oculomotora, anormalidades sacádicas, anormalidades de convergência e acomodação, e estrabismo.
A acuidade visual geralmente é preservada.
QComo os nódulos de Lisch mudam com a idade?
A
Os nódulos de Lisch associados à NF1 aumentam de frequência com a idade. Em menores de 3 anos, ocorrem em apenas 5%, mas acima de 21 anos, estão presentes em quase 100%. Em jovens, podem ser difíceis de detectar mesmo com lâmpada de fenda, sendo necessária interpretação considerando a idade.
As características genéticas de cada doença são as seguintes:
NF1: Cerca de 50% são mutações de novo. Penetrância quase completa, mas fenótipos variados. A neurofibromina funciona como RAS-GAP, convertendo GTP-RAS em GDP-RAS2).
NF2: Perda de função da proteína merlin. É um supressor tumoral expresso principalmente em células de Schwann e células meníngeas; sua deficiência leva a schwannomas, meningiomas e ependimomas.
TSC: Cerca de 2/3 dos casos são esporádicos. Mutações no TSC2 representam 75-80% dos casos esporádicos e têm fenótipo mais grave que mutações no TSC11). Hamartina/tuberina inibem diretamente a via mTOR2).
SWS: Esporádica, causada por mutação somática em mosaico no gene GNAQ1). A mancha vinho do porto (PWB) cobrindo toda a área do primeiro ramo do nervo trigêmeo (V1) apresenta alto risco de complicações oftalmológicas e neurológicas.
VHL: Penetrância quase 100%, cerca de 20% são mutações de novo. pVHL está envolvido na ubiquitinação e degradação do HIF-α pelo proteassoma2). Tipo 1 (sem feocromocitoma) cerca de 80%, Tipo 2 (com feocromocitoma) cerca de 20%2).
AT: Herança autossômica recessiva, a quinase ATM está envolvida no reparo de quebras de fita dupla de DNA e na manutenção da estabilidade genômica 1). O risco de tumores malignos (linfáticos) e a radiosensibilidade são acentuadamente elevados.
Os critérios diagnósticos para NF1 (critérios NIH e Sociedade Japonesa de Dermatologia 2008) estabelecem que o diagnóstico definitivo é feito quando dois ou mais dos sete itens a seguir são preenchidos.
6 ou mais manchas café com leite (pré-púbere: diâmetro ≥5 mm, pós-púbere: ≥15 mm)
Os critérios clínicos incluem schwannoma vestibular bilateral como achado característico. O diagnóstico pode ser mais difícil que o NF1 devido à escassez de sintomas cutâneos. O teste genético detecta mais de 90% das mutações. TC e RM confirmam tumores do nervo auditivo bilateral.
O diagnóstico definitivo é feito com 2 achados principais, ou 1 achado principal + 2 ou mais achados menores. A identificação de uma mutação patogênica em TSC1/TSC2 é reconhecida como um critério diagnóstico independente 1).
Para avaliação do astrocitoma retiniano, utiliza-se exame de fundo de olho, angiografia fluoresceínica e OCT. O diagnóstico diferencial mais importante é o retinoblastoma. A diferenciação é possível pela ausência de calcificação na infância, escassez de vasos nutridores e presença de sintomas sistêmicos, mas a distinção do tumor amora pode ser difícil.
Na escala diagnóstica de Roach, o critério é preencher pelo menos dois dos seguintes: nevo flamejante facial, aumento da pressão intraocular e angioma leptomeníngeo 1). ED-OCT e RM são úteis para avaliar angioma coroideu. A TC de crânio mostra calcificações intracorticais cerebrais.
O teste genético pode detectar mutações VHL em quase 100% dos casos e atualmente é o principal método de diagnóstico definitivo. Recomenda-se exame de fundo de olho com dilatação pupilar anualmente a partir de 1 ano de idade. Tumores esféricos vermelho-alaranjados na periferia com vasos dilatados e tortuosos são achados característicos. A angiografia fluoresceínica é útil para detectar hemangioblastomas periféricos pequenos, e a OCT é usada para avaliar lesões pequenas.
No diagnóstico clínico, a tríade de ataxia, telangiectasia conjuntival bulbar e anormalidade dos movimentos oculares é importante. Achados laboratoriais incluem elevação de AFP sérica, elevação de CA125 e deficiência da proteína ATM (Western blot). A telangiectasia conjuntival bulbar é um achado patognomônico desta doença. A RM mostra atrofia cerebelar difusa na fossa posterior (especialmente vérmis e hemisférios) 1).
Nódulos de Lisch: Hamartomas assintomáticos que não requerem tratamento.
Glioma óptico: Em casos progressivos, considera-se a ressecção cirúrgica, mas a função visual é perdida e as complicações pós-operatórias são frequentes. Em casos de infiltração do quiasma, a quimioterapia é indicada.
Neurofibroma plexiforme: A ressecção cirúrgica total é difícil e propensa a recorrência. Em casos progressivos, pode ser necessária a evisceração orbitária. Em casos irressecáveis, a selumetinibe (inibidor de MEK) é usada como novo tratamento 1).
Glaucoma: O manejo segue o tratamento padrão do glaucoma.
Para schwannoma vestibular, a ressecção cirúrgica é a base; em tumores <3 cm, a audição pode ser preservada em 65% dos casos 1). A ceratite de exposição é tratada com lágrimas artificiais, óculos de proteção corneana e tarsorrafia, se necessário.
Tratamento ocular relacionado à esclerose tuberosa (TSC)
Epilepsia: Para crises parciais infantis e espasmos infantis, a vigabatrina é a primeira escolha1). O astrocitoma subependimal de células gigantes (SEGA) é indicação para ressecção neurocirúrgica1).
O tratamento do glaucoma difere conforme o momento de início.
Glaucoma congênito (início precoce): O tratamento cirúrgico é obrigatório. Trabeculotomia e goniotomia são as opções. É necessária atenção cuidadosa ao descolamento e hemorragia coroidais, com risco de complicações maior que o normal devido ao aumento da pressão venosa epiescleral. Há tendência à resistência ao tratamento medicamentoso.
Tipo de início tardio: primeiro, tratamento medicamentoso; se ineficaz, considerar cirurgia.
Tratamento do hemangioma coroideu é selecionado com base na presença ou ausência de alterações exsudativas a partir do seguinte:
Se ocorrerem alterações exsudativas → fotocoagulação
Fotocoagulação ineficaz ou descolamento exsudativo da retina → radioterapia (dose total de cerca de 20 Gray). Pode-se esperar reposicionamento do descolamento bolhoso da retina e redução do hemangioma.
Fotocoagulação (primeira escolha): Usando laser de argônio ou laser de corante, coagular densamente a retina ao redor do hemangioma e os vasos nutridores, em seguida coagular diretamente o tumor. Para tumores com diâmetro ≥2 discos ópticos, coagular primeiro a artéria aferente e a retina circundante, depois coagular diretamente o tumor.
Criocoagulação: Realizada por via transescleral em casos de hemangiomas grandes ou na periferia retiniana onde a fotocoagulação é difícil.
Vitrectomia: Considerada em casos de alterações exsudativas graves, descolamento de retina ou alterações proliferativas.
Não há tratamento específico para telangiectasia conjuntival. Para imunodeficiência, são usados antibióticos profiláticos e imunoglobulina intravenosa (IVIg)1). A ataxia é tratada sintomaticamente.
QQuando deve ser feito o exame para hemangioma retiniano na VHL?
A
Na VHL, recomenda-se exame de fundo de olho com dilatação uma vez por ano a partir de 1 ano de idade. Todos os familiares de pacientes com mutação confirmada no gene VHL também devem realizar exame de fundo de olho, pois o tratamento precoce pode proporcionar um bom prognóstico visual funcional.
Os mecanismos moleculares de cada doença são mostrados abaixo.
NF1 e TSC (Via mTOR)
NF1 (Neurofibromina): Funciona como RAS-GAP. Acelera a conversão de RAS ligado a GTP (forma ativa) para RAS ligado a GDP (forma inativa). Mutações levam à ativação persistente de RAS, desregulação das vias MAP kinase e PI3K-Akt-mTOR, resultando em proliferação celular descontrolada 2).
TSC (Hamartina/Tuberina): Complexo supressor tumoral que inibe diretamente os complexos mTOR 1 e 2. Mutações levam à hiperatividade de mTOR, causando anormalidades no metabolismo energético, síntese de proteínas/lipídios e sobrevivência celular 2). Mutações no TSC2 resultam em fenótipo mais grave do que mutações no TSC1 1).
VHL (via HIF)
pVHL: Componente do complexo E3 ubiquitina ligase (elongina B/C, Cullin 2, RBX1). Sob oxigênio normal, a prolil hidroxilase hidroxila o HIF-α → pVHL reconhece → ubiquitinação → degradação proteassômica.
Mutação VHL: O estado pseudo-hipóxico persistente leva ao acúmulo de HIF-α, formação de heterodímero com HIF-1β, ativação da transcrição de genes relacionados a VEGF, produção de eritrócitos, metabolismo e proliferação celular, resultando na formação de tumores vasculares 2).
NF2 (Merlin) : proteína supressora de tumor expressa em células de Schwann e células leptomeníngeas. Mutações causam schwannomas, meningiomas e ependimomas.
SWS (mutação somática em mosaico do GNAQ) : anormalidade na sinalização transmembrana associada à proteína G. O padrão de expressão na área do nervo trigêmeo, intracraniana e ocular difere conforme o momento e local da mutação no embrião 1). GNAQ também é a base molecular comum para SWS, KTS e PPV3).
AT (ATM quinase): Fator supressor de tumor envolvido no reparo de quebras de fita dupla do DNA e na manutenção da estabilidade genômica. Mutações → comprometimento do reparo do DNA → predisposição ao câncer, aumento acentuado da radiosensibilidade e imunodeficiência1).
Chevalier et al. (2021) investigaram a associação entre facomatoses e tumores endócrinos e relataram que as vias de sinalização (RAS-PI3K-Akt-mTOR, HIF) em NF1, TSC e VHL estão comumente envolvidas no desenvolvimento de tumores endócrinos múltiplos2).
QPor que as facomatoses causam lesões em múltiplos órgãos?
A
A base fisiopatológica comum das facomatoses é uma anormalidade na formação, migração e diferenciação das células da crista neural. As células da crista neural são derivadas do ectoderma e produzem diversas células, como células de Schwann e melanócitos, resultando em lesões em múltiplos órgãos, incluindo nervos, pele e olhos. Além disso, mutações em vias de sinalização comuns, como RAS-mTOR e VHL-HIF, promovem o desenvolvimento de tumores em diferentes órgãos.
7. Pesquisas recentes e perspectivas futuras (relatos em fase de pesquisa)
Um ensaio clínico está sendo conduzido no consórcio INTUITT-NF2 para schwannomas relacionados à NF2 e tumores progressivos, com resultados promissores relatados 1).
Um ensaio de fase 2 para carcinoma de células renais claras associado a VHL e tumores neuroendócrinos pancreáticos está em andamento.
No estudo de Chevalier et al. (2021) com MK6482, foi obtida resposta objetiva em 64% dos tumores neuroendócrinos pancreáticos, e a taxa de sobrevida livre de progressão em 12 meses foi de 98,3% 2).
Inibidor de mTOR everolimo (tumores associados a TSC)
O estudo EXIST para angiomiolipoma renal e SEGA associados a TSC confirmou resposta morfológica, e a aplicação em tumores endócrinos também é esperada 2).
Descobriu-se que a mutação do gene GNAQ é a base molecular comum para SWS, síndrome de Klippel-Trenaunay (KTS) e nevo vascular pigmentado (PPV)3). Nesses casos de sobreposição, há risco de melanoma de coroide, portanto, recomenda-se exame de fundo de olho com dilatação pupilar 1 a 2 vezes ao ano3).
Gama SM, Tamanini JVG, Moraes MPM, Silva TYT, Lima FT, Pedroso JL, et al. A diagnostic approach to neurocutaneous syndromes. Arquivos de neuro-psiquiatria. 2025;83(7):1-14. doi:10.1055/s-0045-1809664. PMID:40562378; PMCID:PMC12196566.
Chevalier B, Dupuis H, Jannin A, Lemaitre M, Do Cao C, Cardot-Bauters C, et al. Phakomatoses and Endocrine Gland Tumors: Noteworthy and (Not so) Rare Associations. Frontiers in endocrinology. 2021;12:678869. doi:10.3389/fendo.2021.678869. PMID:34025587; PMCID:PMC8134657.
Senthilkumar VA, Kohli P, Mishra C, et al. Ocular features in a patient presenting with a rare combination of multiple phakomatoses. BMJ Case Rep. 2022;15:e252746. doi:10.1136/bcr-2022-252746.
Copie o texto do artigo e cole no assistente de IA de sua preferência.
Artigo copiado para a área de transferência
Abra um assistente de IA abaixo e cole o texto copiado na conversa.