โรคเนื้องอกผิวหนังและระบบประสาท (neurocutaneous syndrome) เป็นคำรวมสำหรับกลุ่มโรคแต่กำเนิดที่ทำให้เกิดรอยโรคแฮมาร์โทมาในผิวหนัง ระบบประสาท และดวงตา
โรคที่เป็นตัวแทนมี 6 โรค ได้แก่ NF1 , NF2, โรคทูเบอรัสสเกลอโรซิส, กลุ่มอาการสเตอร์จ-เวเบอร์, โรคฟอนฮิปเพล-ลินเดา และโรคอะแท็กเซียเทแลงเจียกเทเซีย
ใน NF1 พบก้อนลิช (แฮมาร์โทมาของม่านตา ) ในผู้ใหญ่มากกว่า 90% และเป็นอาการทางตาที่สำคัญสำหรับการวินิจฉัย
ใน SWS เกิดต้อหิน ใน 30-70% ของกรณี และจำเป็นต้องผ่าตัดในต้อหิน แต่กำเนิด
ใน VHL เกิดเนื้องอกหลอดเลือดฝอยจอตา ในผู้ป่วยประมาณ 60% และแนะนำให้ตรวจอวัยวะภายในตาด้วยการขยายม่านตา ทุกปีตั้งแต่อายุ 1 ปี
ภาวะแทรกซ้อนทางตาของแต่ละโรคมักดำเนินไปโดยไม่มีอาการ ดังนั้นการจัดการทางตาอย่างสม่ำเสมอจึงเป็นสิ่งจำเป็น
ยาที่มุ่งเป้าระดับโมเลกุล (สารยับยั้ง MEK, สารยับยั้ง mTOR ฯลฯ) ได้ถูกนำมาใช้เป็นมาตรฐานการรักษาหรือการรักษาใหม่สำหรับโรคบางชนิด
โรคกลุ่มฟาโคมาโทซิส (phakomatoses) เป็นคำเรียกรวมของกลุ่มโรคแต่กำเนิดที่มีลักษณะเฉพาะคือรอยโรคชนิดแฮมาร์โทมา (hamartoma) ที่ผิวหนัง ระบบประสาทส่วนกลาง และดวงตา เรียกอีกอย่างว่า กลุ่มอาการทางผิวหนังและระบบประสาท (neurocutaneous syndromes)
การตั้งชื่อโดยจักษุแพทย์ชาวดัตช์ Van der Hoeve มาจากภาษากรีก «phakos» (เลนส์/จุด) เดิมรวมสามโรค: เนื้องอกเส้นประสาท, โรคทูเบอรัส สเกลอโรซิส, และโรคฟอน ฮิปเพล-ลินเดา ต่อมาเพิ่มกลุ่มอาการสเตอร์จ-เวเบอร์ และภาวะเสียการทรงตัวจากหลอดเลือดฝอยขยาย ปัจจุบันมีการอธิบายมากกว่า 60 กลุ่มอาการ
พื้นฐานทางพยาธิวิทยาร่วมคือความผิดปกติในการสร้าง การเคลื่อนย้าย และการแยกตัวของเซลล์ประสาทคริสตา (neural crest cells) เซลล์ประสาทคริสตามาจากเอ็กโทเดิร์มและสร้างเซลล์หลายชนิด เช่น เซลล์ชวานน์และเมลาโนไซต์ ทำให้เกิดรอยโรคในหลายอวัยวะรวมถึงระบบประสาท ผิวหนัง และดวงตา วิถีสัญญาณที่เกี่ยวข้อง ได้แก่ RAS, MAPK/MEK, mTOR, PI3K/AKT, GNAQ และ VHL-HIF
ความถี่ของการเกิดโรคหลัก 6 โรคมีดังนี้:
โรค ความถี่ (1 ในจำนวนคน) NF1 (นิวโรไฟโบรมาโตซิสชนิดที่ 1)3,000-5,000 ทูเบอรัส สเกลอโรซิส (TSC ) 6,000-10,000 กลุ่มอาการสเตอร์จ-เวเบอร์ (SWS) 20,000-50,000 โรคฟอน ฮิปเพล-ลินเดา (VHL) 36,000 โรคนิวโรไฟโบรมาโทซิสชนิดที่ 2 (NF2) 25,000-100,000 โรคเอแทกเซีย เทเลนเจียกเทเซีย (AT) 88,000 ถึงน้อยกว่า 100,000
Q
โรคกลุ่มฟาโคมาโทซิสมีโรคอะไรบ้าง?
A
โรคหลัก 6 ชนิด ได้แก่ NF1 , NF2, วัณโรคเส้นโลหิตตีบ, กลุ่มอาการสเตอร์จ-เวเบอร์, โรคฟอน ฮิปเพล-ลินเดา และภาวะเสียการทรงตัวจากหลอดเลือดฝอยขยายตัว ทั้งหมดเกิดจากการกลายพันธุ์ของยีนและทำให้เกิดรอยโรคในระบบประสาท ผิวหนัง และดวงตา ปัจจุบันมีกลุ่มอาการมากกว่า 60 ชนิดที่จัดอยู่ในกลุ่มโรคเนื้องอกผิวหนังและระบบประสาท
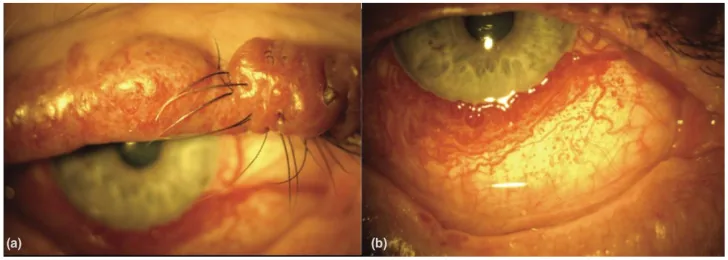
An Update on Multimodal Ophthalmological Imaging of Diffuse Choroidal Hemangioma in Sturge–Weber Syndrome. Vision (Basel). 2023 Oct 6; 7(4):64. Figure 1. PMCID: PMC10594527. License: CC BY.
(a) ปานสีไวน์พอร์ตที่เปลือกตาบนมีก้อนในผู้ป่วย Sturge–Weber syndrome (b) หลอดเลือดเยื่อบุตา กระจายในผู้ป่วย Sturge–Weber syndrome จาก [15].
ชนิดของภาวะแทรกซ้อนทางตาแตกต่างกันในแต่ละโรค และอาการที่ผู้ป่วยรับรู้ก็หลากหลาย
NF1 การมองเห็น ลดลง สูญเสียการมองเห็น สี และข้อบกพร่องของลานสายตา Neurofibroma แบบ plexiform อาจทำให้เกิดตาโปนTuberous sclerosis : Hamartoma เซลล์รูปดาวจอประสาทตา มักไม่มีอาการ ทำให้เกิดความบกพร่องทางการมองเห็น เมื่อกระทบต่อจอประสาทตา หรือจานประสาทตา SWS : เมื่อมีต้อหิน แต่กำเนิดร่วมด้วย จะเกิดกระจกตา ขุ่น น้ำตาไหล และกลัวแสง ในต้อหิน ที่เกิดช้า จะเกิดข้อบกพร่องลานสายตาที่ดำเนินไปโดยไม่เจ็บปวดVHL : ไม่มีอาการในระยะแรก เมื่อ hemangioblastoma ฝอยจอประสาทตา ดำเนินไป จะมีการเปลี่ยนแปลงแบบ exudative, macular edema และวงแหวนสีขาว ทำให้การมองเห็น ลดลงAT : การมองเห็น มักปกติ ความผิดปกติหลักคือความผิดปกติของการเคลื่อนไหวลูกตา (อาตา , apraxia การเคลื่อนไหวลูกตา)
Lisch nodules : อาการแสดงทางตาที่พบบ่อยที่สุดใน NF1 เป็นก้อนเล็กๆ (1-2 มม.) สีน้ำตาลอ่อน ขอบเขตชัด รูปโดม หลายก้อนบนม่านตา ความชุกตามอายุ: <5% ในอายุ <3 ปี, 42% ในอายุ 3-4 ปี, 55% ในอายุ 5-6 ปี, 100% ในอายุ >21 ปี รวมอยู่ในเกณฑ์การวินิจฉัย NF1 (≥2 ก้อน) เนื่องจากม่านตา ของคนญี่ปุ่นเป็นสีน้ำตาล การตรวจด้วย slit-lamp จึงสำคัญเนื้องอกทางเดินประสาทตา (optic glioma) : เกิดขึ้นประมาณ 15-25% ของผู้ป่วย NF1 มักเป็น pilocytic astrocytoma ระดับต่ำ มักไม่มีอาการ เมื่อดำเนินไป จะทำให้ optic atrophy, การมองเห็น บกพร่อง และ visual field defect อาจลุกลามถึง optic chiasmเนื้องอกเส้นประสาทแบบเพล็กซิฟอร์ม : เกิดขึ้นในผู้ป่วย NF1 น้อยกว่า 10% มีลักษณะเฉพาะคือหนังตาผิดรูปเป็นรูปตัว S และเมื่อคลำจะรู้สึกเหมือนถุงไส้เดือน อาจทำให้เกิดตาโปน ตาเหล่ ตาขี้เกียจ และต้อหิน แต่กำเนิดกระดูกปีกใหญ่ของกระดูกสฟีนอยด์เจริญผิดปกติ : ความบกพร่องแต่กำเนิดของผนังเบ้าตา ซึ่งอาจทำให้เกิดตาโปนเป็นจังหวะต้อหิน NF1 ร้อยละ 1-2 มีสองประเภท: แต่กำเนิด (ข้างเดียว) และเกิดช้าอื่นๆ : เส้นประสาทมีเยื่อไมอีลินที่กระจกตา เด่นชัด, คอรอยด์หนา , และเส้นใยประสาทมีเยื่อไมอีลินที่จอตา
เยื่อหุ้มเส้นประสาทตาอักเสบ NF1 ต้อกระจก ต้อกระจก ในวัยหนุ่มสาวแฮมาร์โทมาของจอประสาทตา /อาร์พีอี , เยื่อเหนือจอประสาทตา กระจกตา อักเสบจากการเปิดเปลือกตาไม่สนิทเห็นภาพซ้อน และตาเปิดค้างก้อนลิชพบได้น้อย
แฮมาร์โทมาของเซลล์แอสโทรไซต์ในจอประสาทตา : พบร่วมประมาณ 50% แบ่งเป็น 3 ชนิด: (1) แบบราบ โปร่งแสง ไม่มีหินปูน (2) แบบนูน เป็นก้อนกลม มีหินปูน (ลักษณะคล้ายผลหม่อน) (3) แบบเปลี่ยนผ่าน มักพบหลายก้อนที่ขั้วหลังลูกตารอยโรคจอประสาทตา ที่ไม่มีเม็ดสี : มีลักษณะ “ถูกเจาะทะลุ” (punched-out)ความผิดปกติของหลอดเลือดจอประสาทตา : การโป่งพองคล้ายหลอดเลือดโป่งพองและความผิดปกติของหลอดเลือดแดง-ดำ ซึ่งอาจทำให้เกิดเลือดออกในน้ำวุ้นตา จอประสาทตา ผิดปกติแบบเพิ่มจำนวน และจอประสาทตาลอก อื่นๆ : หลอดเลือดฝอยขยายที่เปลือกตา จุดด่างขาวที่ม่านตา รอยแยกของคอรอยด์ ผิดปกติ
สามอาการหลักคือ (1) หลอดเลือดฝอยขยายบนใบหน้าบริเวณเส้นประสาทไทรเจมินัล (2) หลอดเลือดฝอยขยายในกะโหลกศีรษะข้างเดียวกัน (3) ต้อหิน หรือหลอดเลือดฝอยขยายที่คอรอยด์ ข้างเดียวกัน
ต้อหิน ต้อหิน แต่กำเนิด (แรกเกิดถึง 4 ปี) คิดเป็นประมาณ 60% ทำให้เกิดตาบวม กระจกตา ขุ่น และกระจกตา โต สาเหตุเกี่ยวข้องกับความผิดปกติของมุมลูกตา ความดันหลอดเลือดดำเหนือตาขาว สูง และหลอดเลือดฝอยขยายที่คอรอยด์ มักเกิดร่วมกับหลอดเลือดฝอยขยายที่เปลือกตาHemangioma คอรอยด์ : เกิดขึ้นประมาณ 20-70% ของผู้ป่วย ตรวจพบได้ยากในการตรวจอวัยวะภายในตาปกติเนื่องจากกระจายและขอบเขตไม่ชัดเจน อวัยวะภายในตามีลักษณะคล้ายซอสมะเขือเทศ โดยปกติไม่มีแนวโน้มขยายใหญ่ แต่อาจทำให้เกิดการเปลี่ยนแปลงแบบมีน้ำซึมหรือจอประสาทตาลอก แบบมีน้ำซึมอื่นๆ : การขยายและคดเคี้ยวของหลอดเลือดเยื่อบุตา เอพิสเกลอรา และม่านตา
Hemangioblastoma หลอดเลือดฝอยจอประสาทตา (angioma) : เกิดขึ้นในผู้ป่วย VHL 43-85% (ประมาณ 60% ในรายงานในประเทศ) ประมาณหนึ่งในสามเป็นสองตาและหลายตำแหน่ง พบได้บ่อยบริเวณขมับส่วนปลายของจอประสาทตา เป็นก้อนสีส้มแดงร่วมกับหลอดเลือดนำเข้าและออกที่ขยายและคดเคี้ยว อายุเฉลี่ยที่เริ่มมีอาการคือ 25 ปี มักปรากฏก่อนอายุ 30 ปีเมื่อดำเนินไป จะทำให้เกิดการเปลี่ยนแปลงแบบมีน้ำซึม เลือดออกในจอประสาทตา → จุดรับภาพบวม วงแหวนขาว → การมองเห็น ลดลง
เมื่อดำเนินไปมากขึ้น อาจทำให้เกิดเลือดออกในน้ำวุ้นตา จอประสาทตาลอก แบบดึงรั้ง ต้อหิน ชนิดเส้นเลือดใหม่ → ตาบอด
ในการตรวจหลอดเลือดด้วยฟลูออเรสซีน จะเห็นการไหลของสีเข้าสู่หลอดเลือดแดงนำเข้า → หลอดเลือดดำระบายออก โดยมีการรั่วของสีอย่างชัดเจนจากก้อนเนื้องอกในระยะแรก
หลอดเลือดฝอยขยายที่เยื่อบุตา : อาการทางตาที่พบบ่อยที่สุด มักเกิดขึ้นเมื่ออายุ 5–8 ปี พบในผู้ป่วย 80–90%1) ความผิดปกติของการเคลื่อนไหวตา : อาตา (nystagmus), ภาวะเสียการประสานงานการเคลื่อนไหวลูกตา (oculomotor apraxia), ความผิดปกติของการกลอกตาแบบกระตุก (saccade), ความผิดปกติของการหยีตาและการปรับโฟกัส, และตาเหล่ โดยทั่วไปการมองเห็น ยังคงปกติ
Q
ก้อน Lisch เปลี่ยนแปลงอย่างไรตามอายุ?
A
ก้อน Lisch ที่เกี่ยวข้องกับ NF1 มีความถี่เพิ่มขึ้นตามอายุ ในเด็กอายุต่ำกว่า 3 ปี พบเพียง 5% แต่ในผู้ที่มีอายุมากกว่า 21 ปี พบเกือบ 100% ในวัยเด็กอาจตรวจพบได้ยากแม้ใช้กล้องจุลทรรศน์ชนิดกรีด จึงต้องตีความโดยคำนึงถึงอายุ
รูปแบบการถ่ายทอดทางพันธุกรรม ยีนก่อโรค และตำแหน่งบนโครโมโซมของแต่ละโรคแสดงไว้ด้านล่าง
โรค รูปแบบการถ่ายทอด ยีนก่อโรค ตำแหน่งบนโครโมโซม NF1 ถ่ายทอดแบบออโตโซมเด่น NF1 17q11.2 NF2 ถ่ายทอดแบบออโตโซมเด่น NF2 22q11.1-q13.1 TSC ถ่ายทอดแบบออโตโซมเด่น TSC1/TSC2 9q34/16p13 VHL ถ่ายทอดแบบออโตโซมเด่น VHL 3p25-26 SWS ประปราย (โมเสกโซมาติก) GNAQ 9q21 AT ถ่ายทอดทางออโตโซมแบบด้อย ATM 11q22
ลักษณะทางพันธุกรรมของแต่ละโรคมีดังนี้:
NF1 2) .NF2 : การสูญเสียการทำงานของโปรตีนเมอร์ลิน (merlin) เป็นยีนต้านเนื้องอกที่แสดงออกหลักในเซลล์ชวานน์และเซลล์เยื่อหุ้มสมอง การขาดทำให้เกิดชวานโนมา เมนินจิโอมา และอีเพนไดโมมาTSC 1) ฮามาร์ติน/ทูเบอรีนยับยั้งวิถี mTOR โดยตรง2) .SWS : เกิดแบบประปรายจากการกลายพันธุ์แบบโมเสกในร่างกายของยีน GNAQ1) ปานสีพอร์ตไวน์ (PWB) ที่ครอบคลุมบริเวณสาขาแรกของเส้นประสาทไทรเจมินัล (V1) ทั้งหมดมีความเสี่ยงสูงต่อภาวะแทรกซ้อนทางจักษุวิทยาและระบบประสาทVHL : การแทรกซึมเกือบ 100% ประมาณ 20% เป็นการกลายพันธุ์แบบ de novo pVHL เกี่ยวข้องกับการยูบิควิติเนชันและการย่อยสลาย HIF-α โดยโปรตีอาโซม2) ชนิดที่ 1 (ไม่มีฟีโอโครโมไซโตมา) ประมาณ 80% ชนิดที่ 2 (มีฟีโอโครโมไซโตมา) ประมาณ 20%2) .AT : การถ่ายทอดทางพันธุกรรมแบบออโตโซมด้อย ATM kinase เกี่ยวข้องกับการซ่อมแซมดีเอ็นเอสายคู่ขาดและการรักษาเสถียรภาพของจีโนม 1) ความเสี่ยงของเนื้องอกมะเร็ง (ระบบน้ำเหลือง) และความไวต่อรังสีสูงอย่างมีนัยสำคัญ
ผู้ที่ได้รับการวินิจฉัยว่าเป็นโรคเนื้องอกเนื้อเยื่อประสาทหรือมีสมาชิกในครอบครัวเป็นโรคนี้ ควรตรวจตาเป็นประจำ สำหรับโรค VHL แนะนำให้ตรวจขยายรูม่านตา ทุกปีตั้งแต่อายุ 1 ปี สำหรับโรค SWS การควบคุมความดันลูกตา มีความสำคัญตั้งแต่วัยทารก หากสังเกตเห็นความผิดปกติทางตา (สายตาแย่ลง การเปลี่ยนแปลงของลานตา ตาโปน) ควรปรึกษาแพทย์แต่เนิ่นๆ
เกณฑ์การวินิจฉัย NF1 (เกณฑ์ของ NIH และสมาคมโรคผิวหนังแห่งประเทศญี่ปุ่น พ.ศ. 2551) ระบุว่าการวินิจฉัยที่แน่นอนจะเกิดขึ้นเมื่อมีคุณสมบัติตรงตามเกณฑ์อย่างน้อยสองข้อจากเจ็ดข้อต่อไปนี้
จุดสีน้ำตาลอ่อน (café-au-lait) 6 จุดขึ้นไป (ก่อนวัยเจริญพันธุ์: เส้นผ่านศูนย์กลาง ≥5 มม. หลังวัยเจริญพันธุ์: ≥15 มม.)
เนื้องอกเส้นประสาท (neurofibroma) 2 ชิ้นขึ้นไป หรือเนื้องอกเส้นประสาทแบบกระจาย
จุดกระที่รักแร้หรือขาหนีบ
เนื้องอกเกลียของเส้นประสาทตา (optic glioma)ก้อน Lisch ในม่านตา 2 ก้อนขึ้นไป
รอยโรคกระดูกที่มีลักษณะเฉพาะ (เช่น กระดูกสฟีนอยด์เจริญผิดปกติ)
ญาติสายตรงลำดับที่หนึ่งที่มีภาวะเดียวกัน
บทบาทของการตรวจทางจักษุวิทยามีดังนี้:
กล้องจุลทรรศน์ชนิดกรีดแสง (Slit-lamp) : การตรวจหาก้อนลิช (Lisch nodule) (อาจทำได้ยากในวัยเด็ก)OCT ส่วนหน้าตาและกล้องจุลทรรศน์อัลตราซาวนด์ชีวภาพMRI : วิธีการวินิจฉัยที่ดีที่สุดสำหรับเนื้องอกทางเดินประสาทตา จำเป็นสำหรับการประเมินออปติกไคแอสมาออปติกแทรกต์การตรวจลานสายตา เครื่องตรวจการเชื่อมโยงกันของแสง (OCT ) : การประเมินการบางลงของชั้นใยประสาทจอตาที่เกี่ยวข้องกับเนื้องอกในทางเดินสายตาศักย์ไฟฟ้าสมองที่เกิดจากการกระตุ้นการมองเห็น (VEP ) : การตรวจพบความผิดปกติของเส้นประสาทตา ในระยะเริ่มต้นระยะเวลาการตรวจตามปกติ : มีเพียงก้อนลิช → ปีละ 1 ครั้ง หากมีออปติกไกลโอมา → ทุก 3 เดือน
เกณฑ์ทางคลินิก: ชวานโนมาของเส้นประสาทเวสติบูลาร์ทั้งสองข้างเป็นลักษณะเด่น การวินิจฉัยอาจยากกว่า NF1 เนื่องจากอาการทางผิวหนังน้อย การตรวจทางพันธุกรรมตรวจพบการกลายพันธุ์มากกว่า 90% การตรวจ CT และ MRI เพื่อยืนยันเนื้องอกของเส้นประสาทหูทั้งสองข้าง
การวินิจฉัยที่แน่นอนทำได้โดยมีอาการสำคัญ 2 ข้อ หรืออาการสำคัญ 1 ข้อร่วมกับอาการรอง 2 ข้อขึ้นไป การระบุการกลายพันธุ์ที่ก่อโรคของ TSC1/TSC2 ถือเป็นเกณฑ์การวินิจฉัยที่อิสระ 1) .
ในการประเมิน astrocytoma จอประสาทตา ใช้การตรวจอวัยวะรับภาพ (fundus examination), การถ่ายภาพหลอดเลือดด้วยฟลูออเรสซีน (fluorescein angiography) และ OCT การวินิจฉัยแยกโรคที่สำคัญที่สุดคือ retinoblastoma สามารถแยกได้จากลักษณะที่ไม่มีการกลายเป็นปูนในวัยเด็ก, หลอดเลือดเลี้ยงน้อย, และมีอาการทางระบบ แต่การแยกจาก tumor ชนิด mulberry อาจทำได้ยาก
ตามเกณฑ์การวินิจฉัยของ Roach ต้องมีอย่างน้อย 2 ใน 3 ข้อต่อไปนี้: ปานแดงบนใบหน้า ความดันลูกตา สูง และหลอดเลือดผิดปกติในเยื่อหุ้มสมองชั้นเลปโตเมนิงซ์ 1) การตรวจ ED-OCT และ MRI มีประโยชน์ในการประเมินหลอดเลือดผิดปกติในคอรอยด์ การตรวจ CT ศีรษะพบหินปูนในชั้นคอร์เทกซ์ของสมอง
สำหรับการติดตามโรคต้อหิน จะมีการวัดความดันลูกตา ประเมินเส้นประสาทตา และตรวจลานสายตาเป็นประจำ ในทารกและเด็กเล็ก นอกจากการตรวจด้วยกล้องจุลทรรศน์ชนิดกรีด (slit-lamp) แล้ว อาจจำเป็นต้องตรวจอวัยวะภายในลูกตาและวัดความดันลูกตา ภายใต้การดมยาสลบ
การตรวจทางพันธุกรรมสามารถตรวจพบการกลายพันธุ์ของ VHL ได้เกือบ 100% และปัจจุบันเป็นวิธีการหลักในการวินิจฉัยที่แน่นอน แนะนำให้ตรวจอวัยวะรับภาพโดยขยายม่านตา ทุกปีตั้งแต่อายุ 1 ปี ก้อนกลมสีส้มแดงบริเวณรอบนอกพร้อมหลอดเลือดขยายและคดเคี้ยวเป็นลักษณะเฉพาะ การตรวจหลอดเลือดด้วยฟลูออเรสซีน มีประโยชน์ในการตรวจหา hemangioblastoma ขนาดเล็กบริเวณรอบนอก และ OCT ใช้ในการประเมินรอยโรคขนาดเล็ก
ในการวินิจฉัยทางคลินิก กลุ่มอาการสามอย่างคือ การเคลื่อนไหวไม่ประสานกัน (ataxia) หลอดเลือดฝอยขยายที่เยื่อบุตา (bulbar conjunctival telangiectasia) และความผิดปกติของการเคลื่อนไหวของลูกตาเป็นสิ่งสำคัญ ผลตรวจทางห้องปฏิบัติการพบ AFP ในซีรัมสูงขึ้น CA125 สูงขึ้น และการขาดโปรตีน ATM (Western blot) หลอดเลือดฝอยขยายที่เยื่อบุตา เป็นลักษณะเฉพาะของโรคนี้ (pathognomonic) MRI แสดงการฝ่อของสมองน้อยแบบกระจายในโพรงกะโหลกหลัง (โดยเฉพาะ vermis และ hemisphere) 1)
ก้อน Lisch : เนื้องอกชนิด hamartoma ที่ไม่มีอาการ ไม่จำเป็นต้องรักษาเนื้องอกไกลโอมาของเส้นประสาทตา : ในกรณีที่ลุกลาม พิจารณาการผ่าตัดเอาออก แต่การมองเห็น จะสูญเสียและมีภาวะแทรกซ้อนหลังผ่าตัดมาก ในกรณีที่มีการลุกลามถึงจุดไขว้ประสาทตา การให้เคมีบำบัดเป็นข้อบ่งชี้นิวโรไฟโบรมาแบบเพล็กซิฟอร์ม : การผ่าตัดเอาออกทั้งหมดทำได้ยากและมักกลับเป็นซ้ำ ในกรณีที่ลุกลาม อาจต้องผ่าตัดเอาสิ่งในเบ้าตา ออก ในกรณีที่ไม่สามารถผ่าตัดได้ ใช้เซลูเมตินิบ (ยายับยั้ง MEK) เป็นการรักษาใหม่ 1) ต้อหิน ต้อหิน มาตรฐาน
สำหรับชวานโนมาของเส้นประสาทเวสติบูลาร์ การผ่าตัดเอาออกเป็นพื้นฐาน ในเนื้องอกขนาด <3 ซม. สามารถรักษาการได้ยินได้ใน 65% ของกรณี 1) กระจกตา อักเสบจากการเปิดรับแสงรักษาด้วยน้ำตาเทียม แว่นตาป้องกันกระจกตา และการเย็บเปลือกตาหากจำเป็น
แฮมาร์โทมาสเตลเลตของจอตา : โดยปกติแล้วไม่มีแนวโน้มที่จะขยายใหญ่ขึ้นและไม่จำเป็นต้องรักษาความผิดปกติของหลอดเลือดจอตา (โป่งพอง, ความผิดปกติของหลอดเลือดแดง-ดำ) : ทำการจี้แสงป้องกันเนื่องจากเสี่ยงต่อการตกเลือดในวุ้นตา และจอตาลอก หากเกิดการตกเลือดในวุ้นตา หรือจอตาลอก ให้พิจารณาผ่าตัดวุ้นตา โรคลมชัก : สำหรับอาการชักเฉพาะส่วนในทารกและอาการกระตุกในทารก ยา vigabatrin เป็นทางเลือกแรก1) เนื้องอกแอสโตรไซโตมาชนิดเซลล์ยักษ์ใต้เยื่อบุโพรงสมอง (SEGA) เป็นข้อบ่งชี้ในการผ่าตัดทางประสาทศัลยกรรม1)
การรักษาโรคต้อหิน แตกต่างกันไปตามระยะเวลาที่เริ่มเกิด
โรคต้อหินแต่กำเนิด (เริ่มเกิดเร็ว)คอรอยด์ อย่างเต็มที่ โดยมีความเสี่ยงของภาวะแทรกซ้อนสูงกว่าปกติเนื่องจากความดันหลอดเลือดดำเหนือตาขาว สูงขึ้น มีแนวโน้มที่จะดื้อต่อการรักษาด้วยยาชนิดที่เริ่มมีอาการช้า : ขั้นแรกให้รักษาด้วยยา หากไม่ได้ผลให้พิจารณาการผ่าตัด
การรักษา hemangioma คอรอยด์ เลือกตามว่ามีการเปลี่ยนแปลงแบบ exudative หรือไม่ จากตัวเลือกต่อไปนี้:
หากเกิดการเปลี่ยนแปลงแบบ exudative → การจี้ด้วยแสง
การจี้ด้วยแสง ไม่ได้ผลหรือจอประสาทตาลอก แบบ exudative → การฉายรังสี (ปริมาณรวมประมาณ 20 เกรย์) สามารถคาดหวังให้จอประสาทตาลอก แบบ bullous กลับเข้าที่และ hemangioma หดตัวลงกรณีดื้อต่อการรักษา → การผ่าตัดน้ำวุ้นตา
การรักษาด้วยแสงไดนามิก (PDT ) และการฉายรังสีจากภายนอกก็เป็นทางเลือกได้เช่นกัน3) .
ต้อหิน ใน SWS
โรคต้อหินแต่กำเนิด ที่เกิดร่วมกับ SWS มักควบคุมความดันลูกตา ได้ยากและมักดื้อต่อการรักษาด้วยยาต้อหิน มาตรฐาน ต้องระวังเป็นพิเศษเกี่ยวกับความเสี่ยงของการหลุดลอกของคอรอยด์ และเลือดออกจากความดันหลอดเลือดดำเหนือตาขาว ที่เพิ่มขึ้นระหว่างการผ่าตัด แนะนำให้จัดการในสถานพยาบาลเฉพาะทางตั้งแต่ระยะแรก
หลักการคือการรักษาตั้งแต่ระยะแรก
การจี้ด้วยแสง (ทางเลือกแรก)จอประสาทตา รอบๆ หลอดเลือดผิดปกติและหลอดเลือดเลี้ยงให้แน่นหนา จากนั้นจี้ก้อนเนื้อโดยตรง สำหรับก้อนเนื้อที่มีเส้นผ่านศูนย์กลาง ≥2 หัวประสาทตา ให้จี้หลอดเลือดแดงนำเข้าและจอประสาทตา รอบๆ ก่อน แล้วจึงจี้ก้อนเนื้อโดยตรงการจี้ด้วยความเย็น : ทำผ่านตาขาว ในกรณีหลอดเลือดผิดปกติขนาดใหญ่หรือบริเวณรอบนอกจอประสาทตา ที่การจี้ด้วยแสง ทำได้ยากการผ่าตัดน้ำวุ้นตา จอประสาทตาลอก หรือการเปลี่ยนแปลงแบบ proliferativeระยะสุดท้าย : จำเป็นต้องจัดการกับโรคต้อหิน ชนิด neovascularหากมีประวัติครอบครัว ควรตรวจ fundus ในทุกคนในครอบครัว การรักษาตั้งแต่เนิ่นๆ จะให้ผลการพยากรณ์โรคที่ดี
ไม่มีการรักษาเฉพาะสำหรับ telangiectasia ของเยื่อบุตา สำหรับภาวะภูมิคุ้มกันบกพร่อง ให้ยาปฏิชีวนะป้องกันและการให้อิมมูโนโกลบูลินทางหลอดเลือดดำ (IVIg )1) การรักษาภาวะ ataxia เป็นไปตามอาการ
Q
ควรตรวจคัดกรอง hemangioma จอประสาทตาใน VHL ตั้งแต่เมื่อใด?
A
ในโรค VHL แนะนำให้ตรวจอวัยวะรับภาพ (fundus) โดยขยายม่านตา ปีละครั้งตั้งแต่อายุ 1 ปี สมาชิกในครอบครัวของผู้ป่วยที่ได้รับการยืนยันว่ามีการกลายพันธุ์ของยีน VHL ทุกคนควรได้รับการตรวจอวัยวะรับภาพด้วย เนื่องจากการรักษาตั้งแต่เนิ่นๆ สามารถให้พยากรณ์โรคด้านการทำงานของสายตาที่ดีได้
กลไกการเกิดโรคในระดับโมเลกุลของแต่ละโรคแสดงไว้ด้านล่าง
NF1 และ TSC (เส้นทาง mTOR)
NF1 (นิวโรไฟโบรมิน)2)
TSC (ฮามาร์ติน/ทูเบอริน)2) การกลายพันธุ์ของ TSC2 ทำให้เกิดฟีโนไทป์ที่รุนแรงกว่าการกลายพันธุ์ของ TSC1 1)
VHL (เส้นทาง HIF)
pVHL : ส่วนประกอบของคอมเพล็กซ์ E3 ubiquitin ligase (elongin B/C, Cullin 2, RBX1) ภายใต้ภาวะออกซิเจนปกติ โพรลิลไฮดรอกซีเลสจะไฮดรอกซีเลต HIF-α → pVHL จดจำ → ยูบิควิติเนชัน → การย่อยสลายโดยโปรตีอาโซม
การกลายพันธุ์ของ VHL : ภาวะพร่องออกซิเจนเทียมที่เกิดขึ้นอย่างต่อเนื่องทำให้เกิดการสะสมของ HIF-α การสร้างเฮเทอโรไดเมอร์กับ HIF-1β การกระตุ้นการถอดรหัสของยีนที่เกี่ยวข้องกับ VEGF การสร้างเม็ดเลือดแดง เมแทบอลิซึม และการเพิ่มจำนวนเซลล์ นำไปสู่การเกิดเนื้องอกในหลอดเลือด 2) .
NF2 (เมอร์ลิน) : โปรตีนยับยั้งเนื้องอกที่แสดงออกในเซลล์ชวานน์และเซลล์เยื่อหุ้มสมองชั้นเลปโตเมนิงซ์ การกลายพันธุ์ทำให้เกิดชวานโนมา เมนินจิโอมา และอีเพนไดโมมาSWS (การกลายพันธุ์แบบโมเสกของ GNAQ ในเซลล์ร่างกาย) : ความผิดปกติของการส่งสัญญาณผ่านเยื่อหุ้มเซลล์ที่เกี่ยวข้องกับโปรตีน G รูปแบบการแสดงออกในบริเวณเส้นประสาทไทรเจมินัล ภายในกะโหลกศีรษะ และดวงตาจะแตกต่างกันไปขึ้นอยู่กับช่วงเวลาและตำแหน่งของการกลายพันธุ์ในตัวอ่อน 1) GNAQ ยังเป็นพื้นฐานระดับโมเลกุลร่วมกันของ SWS, KTS และ PPV 3) .AT (ATM kinase) : ปัจจัยยับยั้งเนื้องอกที่เกี่ยวข้องกับการซ่อมแซมดีเอ็นเอสายคู่ขาดและการรักษาเสถียรภาพของจีโนม การกลายพันธุ์ → ความบกพร่องในการซ่อมแซมดีเอ็นเอ → ความไวต่อการเกิดมะเร็ง ความไวต่อรังสีเพิ่มขึ้นอย่างมีนัยสำคัญ และภาวะภูมิคุ้มกันบกพร่อง1)
Chevalier และคณะ (2021) ศึกษาความสัมพันธ์ระหว่างโรคผิวหนังและเนื้องอกต่อมไร้ท่อ และรายงานว่าเส้นทางสัญญาณ (RAS-PI3K-Akt-mTOR, HIF) ใน NF1 , TSC และ VHL มีส่วนร่วมร่วมกันในการเกิดเนื้องอกต่อมไร้ท่อหลายชนิด2) .
Q
ทำไมโรคผิวหนังและเนื้องอกจึงทำให้เกิดรอยโรคในหลายอวัยวะ?
A
พื้นฐานทางพยาธิสรีรวิทยาร่วมของโรคผิวหนังและเนื้องอกคือความผิดปกติในการสร้าง การเคลื่อนที่ และการแยกตัวของเซลล์ประสาทคริสตา เซลล์ประสาทคริสตามาจากเอ็กโทเดิร์มและผลิตเซลล์ต่างๆ เช่น เซลล์ชวานน์และเมลาโนไซต์ ดังนั้นรอยโรคจึงกระจายไปยังหลายอวัยวะรวมถึงเส้นประสาท ผิวหนัง และตา นอกจากนี้ การกลายพันธุ์ในเส้นทางสัญญาณร่วม เช่น RAS-mTOR และ VHL-HIF ยังส่งเสริมการเกิดเนื้องอกข้ามอวัยวะ
เนื้อหาต่อไปนี้อยู่ในขั้นตอนการวิจัยหรือการทดลองทางคลินิกในปัจจุบัน และไม่ใช่การรักษามาตรฐานที่สามารถรับได้ในโรงพยาบาลทั่วไป เป็นข้อมูลอ้างอิงสำหรับผู้เชี่ยวชาญเกี่ยวกับการพัฒนาทางการแพทย์ในอนาคต
ยาที่มุ่งเป้าระดับโมเลกุลที่ได้รับการอนุมัติจาก FDA สำหรับเนื้องอกเส้นประสาทแบบพันกันที่ไม่สามารถผ่าตัดออกได้ มุ่งเป้าไปที่การทำงานเกินปกติของวิถี MAPK ใน NF1 1)
กำลังมีการทดลองทางคลินิกในกลุ่ม INTUITT-NF2 สำหรับชวานโนมาที่เกี่ยวข้องกับ NF2 และเนื้องอกที่ลุกลาม โดยมีรายงานผลลัพธ์ที่มีแนวโน้มดี 1)
การทดลองระยะที่ 2 สำหรับมะเร็งเซลล์ไตชนิด clear cell ที่เกี่ยวข้องกับ VHL และเนื้องอก neuroendocrine ของตับอ่อนกำลังดำเนินอยู่.
ในการทดลอง MK6482 โดย Chevalier และคณะ (2021) พบว่ามีการตอบสนองเชิงวัตถุใน 64% ของเนื้องอกต่อมไร้ท่อของตับอ่อน และอัตราการรอดชีวิตโดยไม่มีการลุกลามที่ 12 เดือนอยู่ที่ 98.3% 2) .
การทดลอง EXIST สำหรับ angiomyolipoma ของไตที่เกี่ยวข้องกับ TSC และ SEGA ยืนยันการตอบสนองทางสัณฐานวิทยา และคาดว่าจะนำไปใช้กับเนื้องอกต่อมไร้ท่อได้เช่นกัน 2) .
พบว่าการกลายพันธุ์ของยีน GNAQ เป็นพื้นฐานระดับโมเลกุลร่วมกันของ SWS, กลุ่มอาการคลิปเปล-เทรนอเนย์ (KTS) และปานหลอดเลือดสี (PPV )3) ในกรณีที่ซ้อนทับกันเหล่านี้ มีความเสี่ยงต่อมะเร็งเมลาโนมาของคอรอยด์ ดังนั้นจึงแนะนำให้ตรวจอวัยวะรับภาพด้วยการขยายม่านตา ปีละ 1-2 ครั้ง3) .
Gama SM, Tamanini JVG, Moraes MPM, Silva TYT, Lima FT, Pedroso JL, et al. A diagnostic approach to neurocutaneous syndromes. Arquivos de neuro-psiquiatria. 2025;83(7):1-14. doi:10.1055/s-0045-1809664. PMID:40562378; PMCID:PMC12196566.
Chevalier B, Dupuis H, Jannin A, Lemaitre M, Do Cao C, Cardot-Bauters C, et al. Phakomatoses and Endocrine Gland Tumors: Noteworthy and (Not so) Rare Associations. Frontiers in endocrinology. 2021;12:678869. doi:10.3389/fendo.2021.678869. PMID:34025587; PMCID:PMC8134657.
Senthilkumar VA, Kohli P, Mishra C, et al. Ocular features in a patient presenting with a rare combination of multiple phakomatoses. BMJ Case Rep. 2022;15:e252746. doi:10.1136/bcr-2022-252746.