Les phacomatoses sont un groupe de maladies congénitales caractérisées par des lésions hamartomateuses de la peau, du système nerveux central et des yeux. Elles sont également appelées syndromes neurocutanés.
La dénomination est due à l’ophtalmologiste néerlandais Van der Hoeve, du grec « phakos » (lentille/tache). Initialement, trois maladies étaient incluses : la neurofibromatose, la sclérose tubéreuse et la maladie de von Hippel-Lindau, auxquelles se sont ajoutés le syndrome de Sturge-Weber et l’ataxie télangiectasique. Actuellement, plus de 60 syndromes sont décrits.
La base pathogénique commune est une anomalie de la formation, de la migration et de la différenciation des cellules de la crête neurale. Les cellules de la crête neurale, dérivées de l’ectoderme, produisent diverses cellules telles que les cellules de Schwann et les mélanocytes, entraînant des lésions dans plusieurs organes : nerfs, peau et yeux. Les voies de signalisation impliquées comprennent RAS, MAPK/MEK, mTOR, PI3K/AKT, GNAQ et VHL-HIF.
Les fréquences d’apparition des six principales maladies sont les suivantes.
QQuelles maladies sont incluses dans les phacomatoses ?
A
Les six maladies représentatives sont la NF1, la NF2, la sclérose tubéreuse, le syndrome de Sturge-Weber, la maladie de von Hippel-Lindau et l’ataxie télangiectasie. Toutes sont dues à des mutations génétiques et provoquent des lésions dans plusieurs organes, notamment les systèmes nerveux, cutané et oculaire. Actuellement, plus de 60 syndromes sont inclus dans la catégorie des phacomatoses.
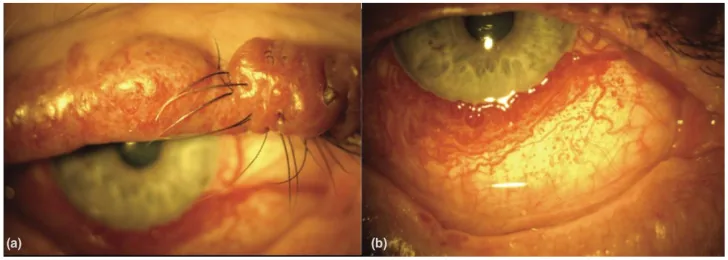
An Update on Multimodal Ophthalmological Imaging of Diffuse Choroidal Hemangioma in Sturge–Weber Syndrome. Vision (Basel). 2023 Oct 6; 7(4):64. Figure 1. PMCID: PMC10594527. License: CC BY.
(a) Tache de vin de Porto de la paupière supérieure avec nodularité chez un patient atteint du syndrome de Sturge-Weber. (b) Vascularisation conjonctivale diffuse chez un patient atteint du syndrome de Sturge-Weber. D’après [15].
Les types de complications oculaires diffèrent selon chaque maladie, et les symptômes subjectifs sont également variés.
NF1 : La progression d’une tumeur du tractus optique (gliome optique) entraîne une baisse de l’acuité visuelle, une perte de la vision des couleurs et des déficits du champ visuel. Le neurofibrome plexiforme peut provoquer une exophtalmie.
Sclérose tubéreuse : L’astrocytome rétinien est généralement asymptomatique. Il peut entraîner une déficience visuelle s’il affecte la macula ou la papille optique.
SWS : En cas de glaucome congénital, il peut y avoir une opacité cornéenne, un larmoiement et une photophobie. Dans le glaucome tardif, un déficit progressif et indolore du champ visuel se produit.
VHL : asymptomatique au début. Lorsque l’hémangiome capillaire rétinien progresse, des modifications exsudatives, un œdème maculaire et des taches blanches annulaires apparaissent, entraînant une baisse de l’acuité visuelle.
AT : l’acuité visuelle est généralement préservée. Les troubles de la motilité oculaire (nystagmus, apraxie oculomotrice) sont prédominants.
Nodules de Lisch : signe oculaire le plus fréquent dans la NF1. Petits nodules (moins de 1 à 2 mm) brun clair, à bords nets, en forme de dôme, multiples sur l’iris. La prévalence selon l’âge augmente avec l’âge : 5 % avant 3 ans, 42 % entre 3 et 4 ans, 55 % entre 5 et 6 ans, 100 % après 21 ans. Ils font partie des critères diagnostiques de la NF1 (≥2). Chez les Japonais, la couleur brune de l’iris rend l’examen à la lampe à fente essentiel.
Tumeur des voies optiques (gliome optique) : survient chez environ 15 à 25 % des patients atteints de NF1. Il s’agit le plus souvent d’un astrocytome pilocytique de bas grade, souvent asymptomatique. En cas de progression, il peut entraîner une atrophie optique, une baisse de l’acuité visuelle et des troubles du champ visuel. Une infiltration chiasmatique est possible.
Neurofibrome plexiforme : survient chez moins de 10 % des patients atteints de NF1. Caractérisé par une « déformation en S » de la paupière, avec une sensation de « sac de vers » à la palpation. Peut provoquer une exophtalmie, un strabisme, une amblyopie et un glaucome congénital.
Dysplasie de l’aile du sphénoïde : défaut congénital de la paroi orbitaire pouvant entraîner une exophtalmie pulsatile.
Glaucome : survient chez 1 à 2 % des patients atteints de NF1. Il existe deux types : congénital (unilatéral) et tardif.
Autres : mise en évidence des nerfs myélinisés cornéens, épaississement choroïdien, nerfs myélinisés rétiniens.
Manifestations oculaires de la NF2 (neurofibromatose de type 2)
Kératite d’exposition : due à une paralysie des nerfs crâniens V et VII causée par un neurinome bilatéral de l’acoustique, entraînant engourdissement facial, diplopie et lagophtalmie.
Les nodules de Lisch sont rares.
Manifestations oculaires de la sclérose tubéreuse de Bourneville (STB)
Hamartome astrocytaire rétinien : présent dans environ 50 % des cas. Classé en 3 types : (i) plat, translucide, non calcifié ; (ii) surélevé, multinodulaire, calcifié (aspect « mûrier ») ; (iii) type transitionnel. Généralement multiples au pôle postérieur.
Anomalies vasculaires rétiniennes : dilatations anévrismales et malformations artérioveineuses, pouvant entraîner une hémorragie du vitré, une rétinopathie proliférante et un décollement de la rétine.
Autres : fibromes angioïdes palpébraux, taches dépigmentées de l’iris, colobome choroïdien atypique.
Manifestations oculaires du SWS (syndrome de Sturge-Weber)
La triade classique comprend (1) un angiome facial dans le territoire du trijumeau, (2) un angiome intracrânien homolatéral, (3) un glaucome ou un angiome choroïdien homolatéral.
Glaucome : manifestation oculaire la plus importante du SWS, survenant dans 30 à 70 % des cas. Le glaucome congénital (de la naissance à 4 ans) représente environ 60 % des cas, entraînant une buphtalmie, une opacité cornéenne et une mégalocornée. L’étiologie implique une dysgénésie de l’angle, une augmentation de la pression veineuse épisclérale et la participation d’un angiome choroïdien. Il survient fréquemment en présence d’un angiome palpébral.
Hémangiome choroïdien : survient dans environ 20 à 70 % des cas. Il est diffus et mal délimité, ce qui le rend difficile à identifier lors d’un examen du fond d’œil normal. Le fond d’œil a un aspect « ketchup ». Il n’a généralement pas tendance à augmenter de taille, mais peut entraîner des modifications exsudatives et un décollement de la rétine exsudatif.
Autres : dilatation et tortuosité des vaisseaux de la conjonctive, de l’épisclère et de l’iris.
Signes oculaires de la maladie de VHL (von Hippel-Lindau)
Hémangiome capillaire rétinien (hémangioblastome) : survient chez 43 à 85 % des patients atteints de VHL (environ 60 % dans les rapports nationaux). Environ un tiers des cas sont bilatéraux et multiples. Il se développe préférentiellement dans la périphérie temporale de la rétine et se présente sous forme de nodules rouge-orange avec des vaisseaux afférents et efférents dilatés et tortueux. L’âge moyen d’apparition est de 25 ans, généralement avant 30 ans.
En progressant, elle entraîne des modifications exsudatives, une hémorragie rétinienne, un œdème maculaire, des taches blanches annulaires et une diminution de l’acuité visuelle.
L’angiographie à la fluorescéine montre un flux de colorant de l’artère afférente vers la veine efférente, avec une fuite précoce et marquée du colorant au niveau de la tumeur.
Manifestations oculaires de l’AT (ataxie télangiectasie)
Télangiectasies conjonctivales : manifestation oculaire la plus fréquente. Surviennent généralement entre 5 et 8 ans et sont présentes chez 80 à 90 % des patients1).
Troubles des mouvements oculaires : nystagmus, apraxie oculomotrice, anomalies des saccades, anomalies de convergence et d’accommodation, strabisme.
L’acuité visuelle est généralement préservée.
QComment les nodules de Lisch évoluent-ils avec l'âge ?
A
Les nodules de Lisch associés à la NF1 augmentent en fréquence avec l’âge. Ils ne sont présents que chez 5 % des enfants de moins de 3 ans, mais on les retrouve chez presque 100 % des personnes de plus de 21 ans. Chez les jeunes patients, ils peuvent être difficiles à détecter même à l’examen à la lampe à fente, et une interprétation tenant compte de l’âge est nécessaire.
Les caractéristiques génétiques de chaque maladie sont les suivantes.
NF1 : environ 50% sont des mutations de novo. Pénétration presque complète mais phénotype variable. La neurofibromine fonctionne comme une RAS-GAP, convertissant le GTP-RAS en GDP-RAS2).
NF2 : perte de fonction de la protéine merline. Suppresseur de tumeur exprimé principalement dans les cellules de Schwann et les cellules méningées, dont la déficience entraîne des schwannomes, méningiomes et épendymomes.
TSC : environ 2/3 des cas sont sporadiques. Les mutations TSC2 représentent 75 à 80% des cas sporadiques et ont un phénotype plus sévère que les mutations TSC11). L’hamartine/tubérine inhibe directement la voie mTOR2).
SWS : sporadique, causée par une mutation somatique en mosaïque du gène GNAQ1). Un angiome plan (PWB) couvrant toute la première branche du trijumeau (V1) présente un risque élevé de complications ophtalmologiques et neurologiques.
VHL : pénétrance presque 100%, environ 20% de mutations de novo. pVHL est impliqué dans l’ubiquitination et la dégradation protéasomique de HIF-α2). Type 1 (sans phéochromocytome) environ 80%, Type 2 (avec phéochromocytome) environ 20%2).
AT : maladie autosomique récessive où la kinase ATM est impliquée dans la réparation des cassures double brin de l’ADN et le maintien de la stabilité génomique 1). Le risque de tumeurs malignes (lymphoïdes) et la radiosensibilité sont considérablement élevés.
Les critères diagnostiques de la NF1 (critères NIH, Société japonaise de dermatologie 2008) établissent le diagnostic définitif lorsque au moins 2 des 7 items suivants sont présents.
6 taches café-au-lait ou plus (avant la puberté : diamètre maximal ≥5 mm ; après la puberté : ≥15 mm)
2 neurofibromes ou plus, ou un neurofibrome plexiforme
Lésions osseuses caractéristiques (par exemple, dysplasie de l’aile du sphénoïde)
Un parent au premier degré atteint de la même maladie
Le rôle de l’examen ophtalmologique est le suivant.
Lampe à fente : Détection des nodules de Lisch (parfois difficile chez les jeunes).
OCT du segment antérieur et microscope ultrasonique : Utiles pour la détection auxiliaire des nodules de Lisch.
IRM : Meilleure méthode de diagnostic des tumeurs des voies optiques. Essentielle pour l’évaluation du chiasma et du tractus optique.
Examen du champ visuel : Dépistage des tumeurs des voies optiques (difficile à réaliser chez les jeunes enfants).
Tomographie par cohérence optique (OCT) : évaluation de l’amincissement de la couche des fibres nerveuses rétiniennes associé aux tumeurs des voies optiques.
PEV (potentiels évoqués visuels) : détection précoce des lésions du nerf optique.
Intervalle d’examen régulier : nodules de Lisch uniquement → 1 fois par an. Gliome du nerf optique présent → 1 fois tous les 3 mois.
Les critères cliniques sont caractérisés par un schwannome vestibulaire bilatéral. Le diagnostic peut être plus difficile que pour la NF1 en raison de la rareté des symptômes cutanés. Les tests génétiques détectent plus de 90 % des mutations. La tomodensitométrie (CT) et l’IRM confirment la présence de neurinomes bilatéraux de l’acoustique.
Diagnostic de la sclérose tubéreuse de Bourneville (TSC)
Le diagnostic est confirmé par la présence de 2 critères majeurs, ou 1 critère majeur et au moins 2 critères mineurs. L’identification d’une mutation pathogène de TSC1/TSC2 est reconnue comme un critère diagnostique indépendant 1).
L’évaluation de l’astrocytome rétinien à cellules géantes utilise l’examen du fond d’œil, l’angiographie à la fluorescéine et l’OCT. Le diagnostic différentiel le plus important est le rétinoblastome. La différenciation est possible grâce à l’absence de calcification, à la pauvreté des vaisseaux nourriciers et à la présence de symptômes systémiques chez l’enfant, mais la distinction avec la tumeur en mûre peut être difficile.
Selon l’échelle diagnostique de Roach, le diagnostic repose sur au moins deux des éléments suivants : angiome plan facial, augmentation de la pression intraoculaire et angiome leptoméningé 1). L’ED-OCT et l’IRM sont utiles pour évaluer l’hémangiome choroïdien. La tomodensitométrie cérébrale montre des calcifications corticales.
La surveillance du glaucome comprend des mesures régulières de la pression intraoculaire, une évaluation du nerf optique et des examens du champ visuel. Chez les nourrissons, en plus de l’examen à la lampe à fente, un examen du fond d’œil et une mesure de la pression intraoculaire sous anesthésie peuvent être nécessaires.
Les tests génétiques détectent des mutations VHL dans près de 100 % des cas et constituent actuellement la méthode de diagnostic de confirmation standard. Un examen du fond d’œil sous dilatation est recommandé une fois par an à partir de l’âge de 1 an. Les tumeurs sphériques rouge-orange de la périphérie associées à des vaisseaux tortueux dilatés sont des signes caractéristiques. L’angiographie à la fluorescéine est utile pour détecter les petits hémangioblastomes périphériques, et l’OCT est utilisée pour évaluer les petites lésions.
Le diagnostic clinique repose sur la triade : ataxie, télangiectasies conjonctivales et anomalies des mouvements oculaires. Les résultats biologiques incluent une élévation de l’AFP sérique, une élévation du CA125 et un déficit en protéine ATM (Western blot). Les télangiectasies conjonctivales sont pathognomoniques de cette maladie. L’IRM montre une atrophie cérébelleuse diffuse de la fosse postérieure (en particulier du vermis et des hémisphères)1).
Nodules de Lisch : hamartomes asymptomatiques ne nécessitant aucun traitement.
Gliome optique : en cas de progression, une résection chirurgicale peut être envisagée, mais la fonction visuelle est perdue et les complications postopératoires sont fréquentes. En cas d’infiltration du chiasma, une chimiothérapie est indiquée.
Neurofibrome plexiforme : l’exérèse chirurgicale totale est difficile et les récidives sont fréquentes. Dans les cas évolués, une énucléation orbitaire peut être nécessaire. Pour les cas non résécables, le sélumétinib (inhibiteur de MEK) est utilisé comme nouveau traitement 1).
Glaucome : prise en charge selon les principes standards du traitement du glaucome.
Pour le schwannome vestibulaire, la résection chirurgicale est la base ; pour les tumeurs de moins de 3 cm, une préservation auditive est possible dans 65 % des cas 1). La kératite d’exposition est traitée par larmes artificielles, lunettes protectrices cornéennes et, si nécessaire, tarsorraphie.
Traitement oculaire de la sclérose tubéreuse (TSC)
Astrocytome hamartomateux rétinien : généralement non évolutif, aucun traitement n’est nécessaire.
Anomalies vasculaires rétiniennes (dilatation anévrismale, malformation artérioveineuse) : photocoagulation prophylactique en raison du risque d’hémorragie du vitré et de décollement de rétine. En cas d’hémorragie du vitré ou de décollement de rétine, envisager une vitrectomie.
Épilepsie : La vigabatrine est considérée comme le traitement de première intention pour les crises partielles et le spasme infantile (syndrome de West) chez les nourrissons1). Le SEGA (astrocytome sous-épendymaire à cellules géantes) est une indication de résection neurochirurgicale1).
Le traitement du glaucome varie en fonction de l’âge d’apparition.
Glaucome congénital (début précoce) : La chirurgie est indispensable. La trabéculotomie et la goniotomie sont les options privilégiées. Une attention particulière doit être portée au décollement choroïdien et aux hémorragies, car le risque de complications est plus élevé que d’habitude en raison de l’augmentation de la pression veineuse épisclérale. La résistance au traitement médicamenteux a tendance à être élevée.
Forme tardive : d’abord un traitement médicamenteux, si inefficace, envisager une chirurgie.
Le traitement de l’hémangiome choroïdien est choisi en fonction de la présence ou non de modifications exsudatives parmi les options suivantes.
En cas de modifications exsudatives → photocoagulation
Photocoagulation inefficace ou décollement de rétine exsudatif → radiothérapie (dose totale d’environ 20 gray). On peut espérer une réapplication du décollement rétinien bulleux et une réduction de l’hémangiome.
Photocoagulation (première intention) : Utilisation d’un laser argon ou dye, coagulation dense de la rétine autour de l’hémangiome et des vaisseaux nourriciers, puis coagulation directe de la tumeur. Pour les tumeurs de plus de 2 diamètres papillaires, coaguler l’artère afférente et la rétine environnante avant de coaguler directement la tumeur.
Cryocoagulation : Réalisée par voie transsclérale lorsque la photocoagulation est difficile en raison de la localisation périphérique du fond d’œil ou d’un hémangiome géant.
Vitrectomie : envisagée en cas de modifications exsudatives sévères, de décollement de la rétine ou de modifications prolifératives.
En cas d’antécédents familiaux, effectuer un examen du fond d’œil pour tous les membres de la famille ; un traitement précoce peut permettre un bon pronostic.
Il n’existe pas de traitement spécifique pour la télangiectasie conjonctivale. Pour l’immunodéficience, des antibiotiques prophylactiques et des immunoglobulines intraveineuses (IVIg) sont administrés 1). L’ataxie est principalement traitée de manière symptomatique.
QÀ partir de quand faut-il commencer le dépistage des hémangiomes rétiniens dans la maladie de VHL ?
A
Dans la VHL, un examen du fond d’œil sous dilatation annuel est recommandé à partir de 1 an. Il est important que tous les membres de la famille des patients présentant une mutation du gène VHL subissent également un examen du fond d’œil, car un traitement précoce permet d’espérer un bon pronostic visuel.
Les mécanismes moléculaires de chaque maladie sont présentés ci-dessous.
NF1 et TSC (voie mTOR)
NF1 (neurofibromine) : agit comme une RAS-GAP. Elle favorise la conversion du RAS lié au GTP (forme active) en RAS lié au GDP (forme inactive). Une mutation entraîne une activation persistante de RAS, une dérégulation des voies MAP kinase et PI3K-Akt-mTOR, et une prolifération cellulaire incontrôlée2).
TSC (hamartine/tubérine) : complexe suppresseur de tumeur qui inhibe directement les complexes mTOR 1 et 2. Une mutation entraîne une hyperactivation de mTOR, des anomalies du métabolisme énergétique, de la synthèse des protéines/lipides et de la survie cellulaire2). Les mutations de TSC2 entraînent un phénotype plus sévère que celles de TSC11).
VHL (voie HIF)
pVHL : composant du complexe E3 ubiquitine ligase (elongin B/C, Cullin 2, RBX1). En condition normoxique, la prolyl hydroxylase hydroxyle HIF-α → pVHL le reconnaît → ubiquitination → dégradation par le protéasome.
Mutation VHL : un état pseudo-hypoxique se produit de manière constante → accumulation de HIF-α → formation d’hétérodimères avec HIF-1β → activation transcriptionnelle des gènes liés au VEGF, à la production de globules rouges, au métabolisme et à la prolifération cellulaire → formation de tumeurs vasculaires2).
NF2 (merline) : protéine suppresseur de tumeur exprimée dans les cellules de Schwann et les cellules leptoméningées. Les mutations entraînent des schwannomes, des méningiomes et des épendymomes.
SWS (mutation somatique mosaïque de GNAQ) : anomalie de la signalisation transmembranaire liée aux protéines G. Le moment et le lieu de la mutation dans l’embryon déterminent les schémas d’expression dans le territoire du nerf trijumeau, le crâne et l’œil1). GNAQ est également la base moléculaire commune à SWS, KTS et PPV3).
AT (kinase ATM) : facteur suppresseur de tumeur impliqué dans la réparation des cassures double brin de l’ADN et le maintien de la stabilité génomique. Mutation → altération de la réparation de l’ADN → prédisposition au cancer, sensibilité accrue aux radiations, immunodéficience1).
Chevalier et al. (2021) ont examiné l’association entre les phacomatoses et les tumeurs endocriniennes, et ont rapporté que les voies de signalisation de NF1, TSC et VHL (voie RAS-PI3K-Akt-mTOR, voie HIF) sont impliquées de manière commune dans le développement des tumeurs endocriniennes multiples 2).
QPourquoi les phacomatoses provoquent-elles des lésions dans plusieurs organes ?
A
La base physiopathologique commune des phacomatoses est une anomalie de la formation, de la migration et de la différenciation des cellules de la crête neurale. Les cellules de la crête neurale, dérivées de l’ectoderme, produisent diverses cellules telles que les cellules de Schwann et les mélanocytes, ce qui entraîne des lésions dans plusieurs organes : nerveux, cutané et oculaire. De plus, les mutations des voies de signalisation communes telles que la voie RAS-mTOR et la voie VHL-HIF favorisent le développement de tumeurs au-delà des organes.
7. Recherches récentes et perspectives futures (rapports en phase de recherche)
C’est un médicament ciblé approuvé par la FDA pour les neurofibromes plexiformes non résécables. Il cible l’hyperactivité de la voie MAPK dans la NF11).
Des essais cliniques sont menés par le consortium INTUITT-NF2 pour les schwannomes associés à la NF2 et les tumeurs progressives, avec des résultats prometteurs rapportés1).
Un essai de phase 2 est en cours pour le carcinome rénal à cellules claires et les tumeurs neuroendocrines pancréatiques associés à VHL.
L’essai de MK6482 par Chevalier et al. (2021) a rapporté un taux de réponse objective de 64 % dans les tumeurs neuroendocrines pancréatiques, avec un taux de survie sans progression à 12 mois de 98,3 % 2).
Inhibiteur mTOR évérolimus (tumeurs associées à la sclérose tubéreuse)
L’essai EXIST sur l’angiomyolipome rénal et le SEGA associés à la TSC a confirmé une réponse morphologique, et son application aux tumeurs endocriniennes est également attendue 2).
Il a été découvert que la mutation du gène GNAQ est une base moléculaire commune au syndrome de Sturge-Weber (SWS), au syndrome de Klippel-Trénaunay (KTS) et à la phacomatose pigmentovasculaire (PPV)3). En raison du risque de mélanome choroïdien dans ces cas de chevauchement, un examen du fond d’œil sous dilatation est recommandé une à deux fois par an3).
Gama SM, Tamanini JVG, Moraes MPM, Silva TYT, Lima FT, Pedroso JL, et al. A diagnostic approach to neurocutaneous syndromes. Arquivos de neuro-psiquiatria. 2025;83(7):1-14. doi:10.1055/s-0045-1809664. PMID:40562378; PMCID:PMC12196566.
Chevalier B, Dupuis H, Jannin A, Lemaitre M, Do Cao C, Cardot-Bauters C, et al. Phakomatoses and Endocrine Gland Tumors: Noteworthy and (Not so) Rare Associations. Frontiers in endocrinology. 2021;12:678869. doi:10.3389/fendo.2021.678869. PMID:34025587; PMCID:PMC8134657.
Senthilkumar VA, Kohli P, Mishra C, et al. Ocular features in a patient presenting with a rare combination of multiple phakomatoses. BMJ Case Rep. 2022;15:e252746. doi:10.1136/bcr-2022-252746.
Copiez le texte de l'article et collez-le dans l'assistant IA de votre choix.
Article copié dans le presse-papiers
Ouvrez un assistant IA ci-dessous et collez le texte copié dans la conversation.