Phakomatosen sind eine Gruppe angeborener Erkrankungen, die durch hamartomatöse Läsionen der Haut, des zentralen Nervensystems und der Augen gekennzeichnet sind. Sie werden auch als neurokutane Syndrome bezeichnet.
Die Benennung geht auf den niederländischen Augenarzt Van der Hoeve zurück und leitet sich vom griechischen Wort „phakos“ (Linse/Fleck) ab. Ursprünglich umfasste es drei Erkrankungen: Neurofibromatose, tuberöse Sklerose und von-Hippel-Lindau-Krankheit, später kamen das Sturge-Weber-Syndrom und die Ataxia teleangiectatica hinzu. Heute sind über 60 Syndrome beschrieben.
Die gemeinsame pathologische Grundlage ist eine Anomalie der Bildung, Migration und Differenzierung von Neuralleistenzellen. Neuralleistenzellen stammen aus dem Ektoderm und produzieren verschiedene Zellen wie Schwann-Zellen und Melanozyten, was zu Läsionen in mehreren Organen (Nerven, Haut, Augen) führt. Beteiligte Signalwege sind RAS, MAPK/MEK, mTOR, PI3K/AKT, GNAQ und VHL-HIF.
Die Häufigkeiten der sechs Haupterkrankungen sind wie folgt.
QWelche Erkrankungen fallen unter die Phakomatosen?
A
Die sechs repräsentativen Erkrankungen sind NF1, NF2, tuberöse Sklerose, Sturge-Weber-Syndrom, von-Hippel-Lindau-Krankheit und Ataxia teleangiectatica. Alle beruhen auf Genmutationen und verursachen Läsionen in mehreren Organen, darunter Nerven-, Haut- und Augensystem. Derzeit werden über 60 Syndrome zur Kategorie der Phakomatosen gezählt.
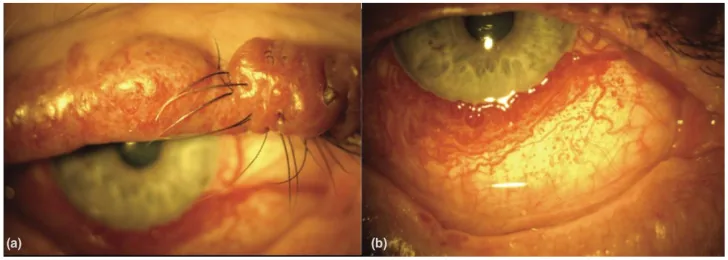
An Update on Multimodal Ophthalmological Imaging of Diffuse Choroidal Hemangioma in Sturge–Weber Syndrome. Vision (Basel). 2023 Oct 6; 7(4):64. Figure 1. PMCID: PMC10594527. License: CC BY.
(a) Portweinfleck des Oberlids mit Knotenbildung bei einem Patienten mit Sturge-Weber-Syndrom. (b) Diffuse konjunktivale Vaskularität bei einem Patienten mit Sturge-Weber-Syndrom. Aus [15].
Die Art der Augenkomplikationen unterscheidet sich je nach Erkrankung, und auch die subjektiven Symptome sind vielfältig.
NF1 : Bei Fortschreiten eines Sehbahntumors (Optikusgliom) treten Sehverschlechterung, Farbsehverlust und Gesichtsfeldausfälle auf. Ein plexiformes Neurofibrom kann zu Exophthalmus führen.
Tuberöse Sklerose : Ein retinales Astrozytom-Hamartom ist in der Regel asymptomatisch. Wenn es die Makula oder die Papille betrifft, kann es zu Sehstörungen kommen.
SWS : Bei begleitendem angeborenem Glaukom treten Hornhauttrübung, Tränenfluss und Photophobie auf. Bei spät einsetzendem Glaukom kommt es zu schmerzlosen, fortschreitenden Gesichtsfeldausfällen.
VHL: Früh asymptomatisch. Bei Fortschreiten des retinalen Kapillarhämangioms treten exsudative Veränderungen, Makulaödem und zirkuläre weiße Flecken auf, die zu Sehverschlechterung führen.
AT: Das Sehvermögen bleibt in der Regel erhalten. Augenbewegungsstörungen (Nystagmus, okulomotorische Apraxie) stehen im Vordergrund.
Lisch-Knötchen: Häufigster Augenbefund bei NF1. Hellbraune, scharf begrenzte, kuppelförmige kleine Knötchen (<1–2 mm) multipel auf der Iris. Die altersabhängige Prävalenz steigt mit dem Alter: <3 Jahre 5 %, 3–4 Jahre 42 %, 5–6 Jahre 55 %, ≥21 Jahre 100 %; sie sind Teil der NF1-Diagnosekriterien (≥2). Bei Japanern ist aufgrund der braunen Irisfarbe eine sorgfältige Spaltlampenuntersuchung wichtig.
Sehnerventumor (Optikusgliom): Tritt bei etwa 15–25 % der NF1-Patienten auf. Meist niedriggradiges pilozytisches Astrozytom, oft asymptomatisch. Bei Progression können Optikusatrophie, Sehverschlechterung und Gesichtsfeldausfälle auftreten. Auch Chiasmainfiltration möglich.
Plexiformes Neurofibrom: Tritt bei weniger als 10 % der NF1-Patienten auf. Charakteristisch ist die „S-förmige Deformität“ des Augenlids mit einem Gefühl wie ein „Wurmsack“ bei der Palpation. Kann Exophthalmus, Strabismus, Amblyopie und angeborenes Glaukom verursachen.
Keilbeinflügeldysplasie: Angeborener Defekt der Orbitawand, der zu einem pulsierenden Exophthalmus führen kann.
Glaukom: Tritt bei 1–2 % der NF1-Patienten auf. Es gibt zwei Typen: angeboren (einseitig) und spät einsetzend.
Sonstiges: Hervortreten von markhaltigen Hornhautnerven, Aderhautverdickung, markhaltige Netzhautnerven.
Expositionskeratitis: sekundär zu beidseitigen Akustikusneurinomen, die zu einer Schädigung der Hirnnerven V und VII führen, was Gesichtstaubheit, Doppelbilder und Lagophthalmus verursacht.
Retinales astrozytäres Hamartom: bei etwa 50 % assoziiert. Drei Typen: (i) flach, durchscheinend, nicht verkalkt; (ii) erhaben, multinodulär, verkalkt (maulbeerartiges Aussehen); (iii) Übergangstyp. Meist multipel am hinteren Pol.
Retinale Gefäßanomalien : aneurysmatische Erweiterungen und arteriovenöse Malformationen, die zu Glaskörperblutung, proliferativer Vitreoretinopathie und Netzhautablösung führen können.
Die Trias besteht aus (1) fazialem Hämangiom im Trigeminusbereich, (2) ipsilateralem intrakraniellem Hämangiom und (3) ipsilateralem Glaukom oder Aderhauthämangiom.
Glaukom : wichtigster Augenbefund bei SWS, tritt bei 30–70 % auf. Angeborenes Glaukom (Geburt bis 4 Jahre) macht etwa 60 % aus und führt zu Buphthalmus, Hornhauttrübung und Megalokornea. Die Ätiologie umfasst Kammerwinkeldysgenesie, erhöhten episkleralen Venendruck und Beteiligung eines Aderhauthämangioms. Es tritt häufig bei Vorliegen eines Lidhämangioms auf.
Aderhaut-Hämangiom: Tritt bei etwa 20–70 % auf. Es ist diffus und unscharf begrenzt, daher bei normaler Fundusuntersuchung schwer zu identifizieren. Der Fundus hat ein „Tomatenketchup“-Aussehen. Es zeigt normalerweise keine Wachstumstendenz, kann aber exsudative Veränderungen und eine exsudative Netzhautablösung verursachen.
Sonstiges : Erweiterung und Schlängelung der Gefäße der Konjunktiva, Episklera und Iris.
Augenbefunde bei VHL (Von-Hippel-Lindau-Krankheit)
Retinales Kapillarhämangiom (Hämangioblastom): Tritt bei 43–85 % der VHL-Patienten auf (in nationalen Berichten etwa 60 %). Etwa ein Drittel der Fälle ist bilateral und multipel. Es tritt bevorzugt in der temporalen Netzhautperipherie auf und wird als rötlich-oranger Knoten mit erweiterten, geschlängelten zu- und abführenden Gefäßen beobachtet. Das durchschnittliche Erkrankungsalter beträgt 25 Jahre, in der Regel tritt es vor dem 30. Lebensjahr auf.
Im Verlauf kommt es zu exsudativen Veränderungen, Netzhautblutungen, Makulaödem, ringförmigen weißen Flecken und Sehverschlechterung.
Bei weiterem Fortschreiten kann es zu Glaskörperblutung, traktiver Netzhautablösung, neovaskulärem Glaukom und schließlich zur Erblindung kommen.
In der Fluoreszenzangiographie zeigt sich ein Farbstofffluss von der zuführenden Arterie zur abführenden Vene, mit früher und deutlicher Farbstoffleckage im Tumorbereich.
Teleangiektasien der Bindehaut : häufigster Augenbefund. Tritt meist im Alter von 5–8 Jahren auf und ist bei 80–90 % der Patienten vorhanden1).
Augenbewegungsstörungen : Nystagmus, okulomotorische Apraxie, Sakkadenstörungen, Konvergenz- und Akkommodationsstörungen, Strabismus.
Die Sehschärfe bleibt in der Regel erhalten.
QWie verändern sich Lisch-Knötchen mit dem Alter?
A
Lisch-Knötchen bei NF1 treten mit zunehmendem Alter häufiger auf. Bei Kindern unter 3 Jahren sind sie nur in 5 % der Fälle vorhanden, bei Personen über 21 Jahren jedoch nahezu zu 100 %. Bei jungen Patienten können sie selbst bei der Spaltlampenuntersuchung schwer zu erkennen sein, und eine altersgerechte Interpretation ist erforderlich.
Die genetischen Merkmale der einzelnen Erkrankungen sind wie folgt.
NF1: etwa 50% sind De-novo-Mutationen. Nahezu vollständige Penetranz, aber variabler Phänotyp. Neurofibromin fungiert als RAS-GAP und wandelt GTP-RAS in GDP-RAS um2).
NF2: Funktionsverlust des Merlin-Proteins. Tumorsuppressor, der hauptsächlich in Schwann-Zellen und Meningealzellen exprimiert wird; ein Defekt führt zu Schwannomen, Meningeomen und Ependymomen.
TSC: etwa 2/3 der Fälle sind sporadisch. TSC2-Mutationen machen 75–80% der sporadischen Fälle aus und haben einen schwereren Phänotyp als TSC1-Mutationen1). Hamartin/Tuberin hemmt direkt den mTOR-Signalweg2).
SWS: sporadisch, verursacht durch somatische Mosaikmutationen im GNAQ-Gen1). Ein Portwein-Fleck (PWB), der das gesamte Versorgungsgebiet des ersten Trigeminusasts (V1) bedeckt, birgt ein hohes Risiko für ophthalmologische und neurologische Komplikationen.
VHL: Penetranz nahezu 100%, etwa 20% De-novo-Mutationen. pVHL ist an der Ubiquitinierung und dem proteasomalen Abbau von HIF-α beteiligt2). Typ 1 (ohne Phäochromozytom) etwa 80%, Typ 2 (mit Phäochromozytom) etwa 20%2).
AT : autosomal-rezessiv vererbte Erkrankung, bei der die ATM-Kinase an der Reparatur von DNA-Doppelstrangbrüchen und der Aufrechterhaltung der Genomstabilität beteiligt ist 1). Das Risiko für bösartige Tumore (lymphatisch) und die Strahlenempfindlichkeit sind deutlich erhöht.
Die Diagnosekriterien der NF1 (NIH-Kriterien, Japanische Dermatologische Gesellschaft 2008) besagen, dass die definitive Diagnose gestellt wird, wenn mindestens 2 der folgenden 7 Kriterien erfüllt sind.
6 oder mehr Café-au-lait-Flecken (vor der Pubertät: maximaler Durchmesser ≥5 mm; nach der Pubertät: ≥15 mm)
2 oder mehr Neurofibrome oder ein plexiformes Neurofibrom
Sommersprossen in den Achselhöhlen oder der Leiste
Klinische Kriterien sind durch bilaterale Vestibularisschwannome gekennzeichnet. Aufgrund der geringen Hautsymptome kann die Diagnose schwieriger sein als bei NF1. Gentests erkennen über 90 % der Mutationen. CT und MRT bestätigen bilaterale Akustikusneurinome.
Die Diagnose wird durch 2 Hauptkriterien oder 1 Hauptkriterium + 2 oder mehr Nebenkriterien bestätigt. Der Nachweis einer pathogenen TSC1/TSC2-Mutation gilt als unabhängiges Diagnosekriterium 1).
Zur Beurteilung des retinalen Astrozytenhamartoms werden Funduskopie, Fluoreszenzangiographie und OCT eingesetzt. Die wichtigste Differentialdiagnose ist das Retinoblastom. Die Unterscheidung ist möglich durch fehlende Verkalkung, spärliche versorgende Gefäße und systemische Symptome im Kindesalter, aber die Abgrenzung zum Maulbeertumor kann schwierig sein.
Nach der Roach-Diagnoseskala müssen mindestens zwei der folgenden Kriterien erfüllt sein: Gesichts-Portweinfleck, erhöhter Augeninnendruck und leptomeningeales Angiom 1). ED-OCT und MRT sind zur Beurteilung des Aderhautangioms nützlich. Im CT des Kopfes zeigen sich Verkalkungen in der Hirnrinde.
Das Glaukom-Monitoring umfasst regelmäßige Augeninnendruckmessungen, Beurteilung des Sehnervs und Gesichtsfelduntersuchungen. Bei Säuglingen kann zusätzlich zur Spaltlampenuntersuchung eine Funduskopie und Augeninnendruckmessung unter Narkose erforderlich sein.
Gentests können in fast 100 % der Fälle VHL-Mutationen nachweisen und sind derzeit die Standardmethode zur definitiven Diagnose. Eine Fundusuntersuchung mit Pupillenerweiterung wird ab dem 1. Lebensjahr einmal jährlich empfohlen. Rötlich-orange kugelförmige Tumoren in der Peripherie mit erweiterten, geschlängelten Gefäßen sind charakteristische Befunde. Die Fluoreszenzangiographie ist nützlich zur Erkennung kleiner peripherer Hämangioblastome, und die OCT wird zur Beurteilung kleiner Läsionen eingesetzt.
Für die klinische Diagnose ist die Trias aus Ataxie, konjunktivalen Teleangiektasien und Augenbewegungsstörungen wichtig. Erhöhtes Serum-AFP, erhöhtes CA125 und ATM-Proteinmangel (Western Blot) sind Laborbefunde. Konjunktivale Teleangiektasien sind pathognomonisch für diese Erkrankung. Die MRT zeigt eine diffuse Kleinhirnatrophie der hinteren Schädelgrube (insbesondere des Vermis und der Hemisphären)1).
Lisch-Knötchen: asymptomatische Hamartome, die keiner Behandlung bedürfen.
Optikusgliom : Bei Progredienz wird eine chirurgische Resektion erwogen, jedoch geht die Sehfunktion verloren und postoperative Komplikationen sind häufig. Bei Infiltration des Chiasmas ist eine Chemotherapie indiziert.
Plexiformes Neurofibrom : Die vollständige chirurgische Entfernung ist schwierig und Rezidive sind häufig. In fortgeschrittenen Fällen kann eine Orbitaeviszeration erforderlich sein. Bei nicht resezierbaren Fällen wird Selumetinib (MEK-Inhibitor) als neue Therapie eingesetzt 1).
Glaukom : Behandlung gemäß den üblichen Glaukomtherapierichtlinien.
Bei Vestibularisschwannomen ist die chirurgische Resektion die Grundlage; bei Tumoren unter 3 cm ist in 65 % der Fälle ein Hörerhalt möglich 1). Die Expositionskeratitis wird mit künstlichen Tränen, Hornhautschutzbrillen und ggf. Tarsorrhaphie behandelt.
Augenbezogene Behandlung der tuberösen Sklerose (TSC)
Retinales Astrozytom-Hamartom : In der Regel zeigt es keine Wachstumstendenz und bedarf keiner Behandlung.
Netzhautgefäßanomalien (aneurysmatische Erweiterung, arteriovenöse Malformation) : prophylaktische Photokoagulation aufgrund des Risikos von Glaskörperblutung und Netzhautablösung. Bei Auftreten von Glaskörperblutung oder Netzhautablösung ist eine Vitrektomie in Betracht zu ziehen.
Epilepsie: Vigabatrin gilt als Mittel der ersten Wahl bei fokalen Anfällen und infantilen Spasmen (West-Syndrom) bei Säuglingen1). SEGA (subependymales Riesenzellastrozytom) ist eine Indikation für die neurochirurgische Resektion1).
Die Behandlung des Glaukoms richtet sich nach dem Zeitpunkt des Auftretens.
Angeborenes Glaukom (früher Beginn) : Eine chirurgische Behandlung ist zwingend erforderlich. Trabekulotomie und Goniotomie werden bevorzugt. Auf Aderhautablösung und Blutungen ist besonders zu achten, da das Komplikationsrisiko aufgrund des erhöhten episkleralen Venendrucks höher als üblich ist. Die Resistenz gegenüber medikamentöser Behandlung ist tendenziell hoch.
Spätmanifestation : Zunächst medikamentöse Behandlung, bei Unwirksamkeit Operation in Betracht ziehen.
Die Behandlung des Aderhautangioms wird je nach Vorhandensein exsudativer Veränderungen aus den folgenden Optionen ausgewählt.
Bei exsudativen Veränderungen → Photokoagulation
Photokoagulation unwirksam oder exsudative Netzhautablösung → Strahlentherapie (Gesamtdosis etwa 20 Gray). Eine Wiederanlegung der bullösen Netzhautablösung und Verkleinerung des Angioms sind zu erwarten.
Photokoagulation (erste Wahl) : Verwendung eines Argon- oder Farbstofflasers, dichte Koagulation der Netzhaut um das Hämangiom und der versorgenden Gefäße, dann direkte Koagulation des Tumors. Bei Tumoren mit mehr als 2 Papillendurchmessern wird nach Koagulation der zuführenden Arterie und der umgebenden Netzhaut der Tumor direkt koaguliert.
Kryokoagulation : Transskleral durchgeführt, wenn die Photokoagulation aufgrund einer peripheren Funduslage oder eines riesigen Hämangioms schwierig ist.
Vitrektomie : wird bei starken exsudativen Veränderungen, Netzhautablösung oder proliferativen Veränderungen in Betracht gezogen.
Endstadium : Management des Neovaskularisationsglaukoms erforderlich.
Bei positiver Familienanamnese sollte bei allen Familienmitgliedern eine Fundusuntersuchung durchgeführt werden; eine frühzeitige Behandlung verspricht eine gute Prognose.
Es gibt keine spezifische Behandlung für konjunktivale Teleangiektasien. Bei Immundefizienz werden prophylaktische Antibiotika und intravenöse Immunglobuline (IVIg) verabreicht 1). Die Ataxie wird hauptsächlich symptomatisch behandelt.
QAb wann sollte man auf Netzhautangiome bei VHL untersuchen?
A
Bei VHL wird ab dem 1. Lebensjahr eine jährliche Fundusuntersuchung in Mydriasis empfohlen. Es ist wichtig, dass auch alle Familienmitglieder von Patienten mit bestätigter VHL-Genmutation eine Fundusuntersuchung erhalten, da eine frühzeitige Behandlung eine gute visuelle Prognose erwarten lässt.
Die molekularen Pathogenesemechanismen der einzelnen Erkrankungen sind im Folgenden dargestellt.
NF1 und TSC (mTOR-Signalweg)
NF1 (Neurofibromin) : fungiert als RAS-GAP. Es fördert die Umwandlung von GTP-gebundenem RAS (aktiv) in GDP-gebundenes RAS (inaktiv). Mutation führt zu persistierender RAS-Aktivierung, Deregulierung des MAP-Kinase- und des PI3K-Akt-mTOR-Signalwegs sowie unkontrollierter Zellproliferation2).
TSC (Hamartin/Tuberin) : Tumorsuppressorkomplex, der direkt die mTOR-Komplexe 1 und 2 hemmt. Mutation führt zu mTOR-Überaktivierung, Anomalien im Energiestoffwechsel, der Protein-/Lipidsynthese und des Zellüberlebens2). TSC2-Mutationen führen zu einem schwereren Phänotyp als TSC1-Mutationen1).
VHL (HIF-Signalweg)
pVHL : Bestandteil des E3-Ubiquitin-Ligase-Komplexes (Elongin B/C, Cullin 2, RBX1). Unter Normoxie hydroxyliert Prolylhydroxylase HIF-α → pVHL erkennt es → Ubiquitinierung → proteasomaler Abbau.
VHL-Mutation : Es entsteht ständig ein pseudohypoxischer Zustand → HIF-α-Akkumulation → Heterodimerbildung mit HIF-1β → transkriptionelle Aktivierung von Genen, die mit VEGF, Erythrozytenproduktion, Stoffwechsel und Zellproliferation zusammenhängen → Bildung von Gefäßtumoren2).
NF2 (Merlin) : Tumorsuppressorprotein, das in Schwann-Zellen und Leptomeninx-Zellen exprimiert wird. Mutationen führen zu Schwannomen, Meningeomen und Ependymomen.
SWS (GNAQ somatische Mosaikmutation) : Anomalie der G-Protein-vermittelten Transmembransignalübertragung. Zeitpunkt und Ort der Mutation im Embryo bestimmen die Expressionsmuster im Trigeminusbereich, intrakraniell und im Auge1). GNAQ ist auch die gemeinsame molekulare Grundlage für SWS, KTS und PPV3).
AT (ATM-Kinase) : Tumorsuppressor, der an der Reparatur von DNA-Doppelstrangbrüchen und der Aufrechterhaltung der Genomstabilität beteiligt ist. Mutation → gestörte DNA-Reparatur → Krebsprädisposition, stark erhöhte Strahlenempfindlichkeit, Immundefizienz1).
Chevalier et al. (2021) untersuchten den Zusammenhang zwischen Phakomatosen und endokrinen Tumoren und berichteten, dass die Signalwege von NF1, TSC und VHL (RAS-PI3K-Akt-mTOR-Weg, HIF-Weg) gemeinsam an der Entstehung multipler endokriner Tumoren beteiligt sind 2).
QWarum verursachen Phakomatosen Läsionen in mehreren Organen?
A
Die gemeinsame pathophysiologische Grundlage der Phakomatosen ist eine Störung der Bildung, Migration und Differenzierung von Neuralleistenzellen. Neuralleistenzellen stammen aus dem Ektoderm und produzieren verschiedene Zellen wie Schwann-Zellen und Melanozyten, was zu Läsionen in mehreren Organen (Nerven, Haut, Auge) führt. Darüber hinaus fördern Mutationen in gemeinsamen Signalwegen wie dem RAS-mTOR-Weg und dem VHL-HIF-Weg die Tumorentstehung über Organe hinweg.
7. Aktuelle Forschung und Zukunftsperspektiven (Berichte aus der Forschungsphase)
Dies ist ein zielgerichtetes Medikament, das von der FDA für nicht resezierbare plexiforme Neurofibrome zugelassen wurde. Es zielt auf die Überaktivität des MAPK-Signalwegs bei NF1 ab1).
Klinische Studien werden vom INTUITT-NF2-Konsortium für NF2-assoziierte Schwannome und fortschreitende Tumore durchgeführt, mit vielversprechenden Ergebnissen1).
Eine Phase-2-Studie für VHL-assoziierte klarzellige Nierenzellkarzinome und pankreatische neuroendokrine Tumoren läuft derzeit.
Die Studie von Chevalier et al. (2021) zu MK6482 zeigte eine objektive Ansprechrate von 64 % bei pankreatischen neuroendokrinen Tumoren und eine 12-Monats-Progressionsfreiheitsrate von 98,3 % 2).
Die EXIST-Studie zu TSC-assoziierten renalen Angiomyolipomen und SEGA hat ein morphologisches Ansprechen bestätigt, und eine Anwendung bei endokrinen Tumoren wird ebenfalls erwartet 2).
Es wurde festgestellt, dass die GNAQ-Genmutation eine gemeinsame molekulare Grundlage für das Sturge-Weber-Syndrom (SWS), das Klippel-Trénaunay-Syndrom (KTS) und die Phakomatosis pigmentovascularis (PPV) darstellt3). Aufgrund des Risikos eines Aderhautmelanoms in diesen Überlappungsfällen wird eine jährliche oder zweimal jährliche Fundusuntersuchung in Mydriasis empfohlen3).
Gama SM, Tamanini JVG, Moraes MPM, Silva TYT, Lima FT, Pedroso JL, et al. A diagnostic approach to neurocutaneous syndromes. Arquivos de neuro-psiquiatria. 2025;83(7):1-14. doi:10.1055/s-0045-1809664. PMID:40562378; PMCID:PMC12196566.
Chevalier B, Dupuis H, Jannin A, Lemaitre M, Do Cao C, Cardot-Bauters C, et al. Phakomatoses and Endocrine Gland Tumors: Noteworthy and (Not so) Rare Associations. Frontiers in endocrinology. 2021;12:678869. doi:10.3389/fendo.2021.678869. PMID:34025587; PMCID:PMC8134657.
Senthilkumar VA, Kohli P, Mishra C, et al. Ocular features in a patient presenting with a rare combination of multiple phakomatoses. BMJ Case Rep. 2022;15:e252746. doi:10.1136/bcr-2022-252746.
Kopieren Sie den Artikeltext und fügen Sie ihn in den KI-Assistenten Ihrer Wahl ein.
Artikel in die Zwischenablage kopiert
Öffnen Sie unten einen KI-Assistenten und fügen Sie den kopierten Text in den Chat ein.