Die thrombotisch-thrombozytopenische Purpura (TTP), auch Moschcowitz-Krankheit genannt, ist eine seltene Bluterkrankung. Sie ist durch eine Pentade gekennzeichnet: mikroangiopathische hämolytische Anämie (MAHA), thrombozytopenische Purpura, akute Nierenschädigung, neurologische Auffälligkeiten (Schwankungen des mentalen Zustands) und Fieber.
Ophthalmologische Zeichen werden bei 14–20 % der Fälle berichtet und zeigen vielfältige Befunde, die auf Mikrothromben in den Netzhaut- und Aderhautgefäßen zurückzuführen sind.
Die Inzidenz wird auf 3,7 bis 11 Fälle pro Million Einwohner pro Jahr geschätzt. In der erwachsenen Bevölkerung Frankreichs wird sie mit 1,5 Fällen pro Million Einwohner pro Jahr angegeben5). Die jährliche Prävalenz beträgt etwa 10 Fälle pro Million Einwohner7), wobei Frauen häufiger betroffen sind.
Geschlechterunterschied : Frauen erkranken 2- bis 3-mal häufiger als Männer6).
Begünstigende Faktoren : Häufiger bei Menschen afro-karibischer Herkunft und adipösen Patienten.
Mortalität ohne Behandlung : Etwa 90%. Durch Einführung des Plasmaaustauschs sinkt sie auf 10-20%3).
Die TTP wird in angeborene und erworbene Formen unterteilt.
Angeborene TTP (Upshaw-Schulman-Syndrom) : verursacht durch ADAMTS13-Genmutation. Macht etwa 5 % aller Fälle aus. In der NCBI ClinVar-Datenbank wurden über 260 Mutationsstellen identifiziert, davon etwa 60 % Missense-Mutationen und etwa 20 % kleine Deletionen/Insertionen6).
Erworbene TTP : verursacht durch Autoantikörper gegen ADAMTS13, macht etwa 95 % aller Fälle aus3).
QWie häufig treten ophthalmologische Zeichen der TTP auf?
A
Ophthalmologische Zeichen werden bei 14–20 % der Fälle berichtet. Dazu gehören Netzhautblutungen, Gefäßverschlüsse, seröse Netzhautablösungen und Papillenödem. Augensymptome können den systemischen Symptomen vorausgehen, sodass der Augenarzt möglicherweise als erster eine TTP vermutet.
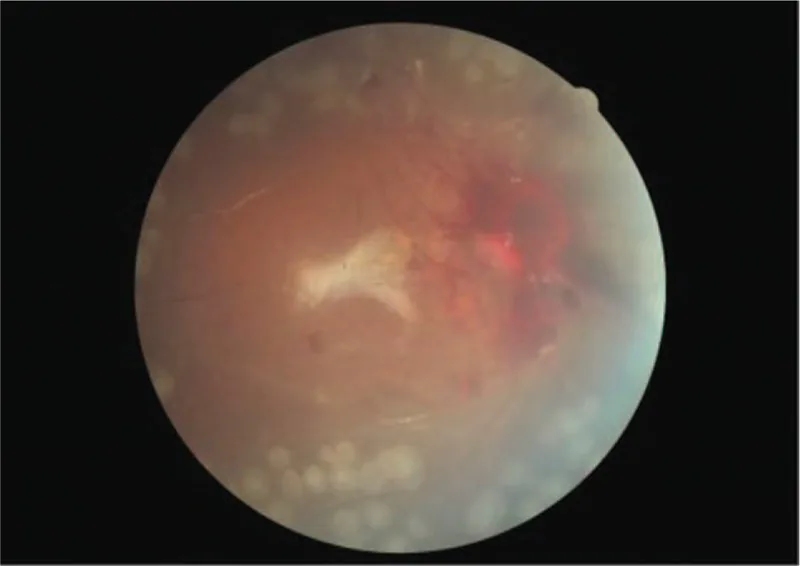
Bilateral proliferative retinopathy and ischemic optic neuropathy in a patient with atypical hemolytic-uremic syndrome: A case report. Medicine (Baltimore). 2019 Sep 27; 98(39):e17232. Figure 4. PMCID: PMC6775429. License: CC BY.
Fundus-Farbfotografie des rechten Auges nach der Operation. Obwohl eine sichtbare Blutung um die Papille herum vorhanden ist, ist die Netzhaut gut anliegend.
Folgende ophthalmologische subjektive Symptome wurden berichtet.
Akuter Sehverlust : aufgrund einer Netzhautläsion.
Verschwommenes Sehen : tritt sekundär zu einem Papillenödem auf.
Diplopie : verbunden mit einer Augenbewegungsstörung infolge einer Hirnnervenlähmung.
Vorübergehendes verschwommenes Sehen und Gelbsehen: Berichtet bei Fällen von Schwangerschafts-TTP3).
Zu den systemischen Symptomen der TTP gehören Kopfschmerzen, Bewusstseinsveränderungen, psychische Störungen, Epilepsie, fokale neurologische Ausfälle, Fieber, Müdigkeit, Gelenkschmerzen, Gelbsucht, Übelkeit und Erbrechen1). Haut- und Schleimhautblutungen (Purpura, Petechien, Zahnfleischbluten) treten sekundär zur Thrombozytopenie auf3).
Die zugrunde liegende Ursache der TTP ist eine verminderte ADAMTS13-Aktivität. ADAMTS13 ist eine Protease, die von-Willebrand-Faktor (vWF)-Riesenmultimere spaltet. Wenn seine Aktivität abnimmt, werden die vWF-Riesenmultimere nicht gespalten und sammeln sich im Blutkreislauf an, wodurch thrombozytenreiche Mikrothromben entstehen.
Die Ursache ist eine ADAMTS13-Genmutation, die häufig durch eine Schwangerschaft ausgelöst wird. Bei der angeborenen TTP beträgt das Rückfallrisiko während der Schwangerschaft 100 %, bei der erworbenen TTP 0–50 % 3).
Impfstoff : Es wurden Fälle nach einer COVID-19-Impfung berichtet, am häufigsten nach BNT162b2 (7 von 10 Fällen in der Literatur) 5).
Chirurgie : Es gibt Fälle nach Herzoperationen (Klappenersatz, TAVR)4).
HIV-Infektion7).
QIst TTP erblich?
A
Die angeborene TTP (Upshaw-Schulman-Syndrom) wird durch Mutationen im ADAMTS13-Gen verursacht und ist eine Erbkrankheit. Die NCBI ClinVar-Datenbank enthält über 260 Mutationsstellen, die häufig in Form von zusammengesetzten heterozygoten Mutationen auftreten. Allerdings sind etwa 95 % aller TTP-Fälle erworben und werden durch Autoantikörper gegen ADAMTS13 verursacht.
Die Diagnose des TTP basiert hauptsächlich auf dem Nachweis einer mikroangiopathischen hämolytischen Anämie (MAHA) und einer thrombozytopenischen Purpura. Die Behandlung muss ohne Verzögerung begonnen werden, ohne die Ergebnisse des ADAMTS13-Aktivitätstests abzuwarten.
Darüber hinaus ist eine Abgrenzung zu Erkrankungen mit niedriger ADAMTS13-Aktivität wie Sepsis, DIC, Lebererkrankungen und Malaria tropica erforderlich.
Wenn ophthalmologische Befunde vorliegen, führen Sie eine detaillierte Befragung zu anderen systemischen Symptomen durch und prüfen Sie das Vorliegen der klassischen 5 Zeichen des TTP.
Der Plasmaaustausch ist die Erstlinientherapie. Es werden vier Wirkungen erwartet.
Zufuhr von ADAMTS13
Entfernung von Inhibitoren (Autoantikörpern)
Entfernung von ultrahochmolekularen vWF-Multimeren (UL-vWFM)
Zufuhr von normalem vWF
Bei Schwangerschafts-TTP werden 2.000 ml/Sitzung (40-60 ml/kg) 1-2 mal täglich durchgeführt 3). Die unbehandelte Sterblichkeitsrate von 90 % kann durch Einführung des Plasmaaustauschs auf 10-20 % gesenkt werden 3).
Bei fehlendem schwerem erworbenem ADAMTS13-Mangel ist auch die Transfusion von gefrorenem Frischplasma eine Option.
Wird als adjuvante Therapie bei therapierefraktären oder rezidivierenden Fällen eingesetzt.
Standarddosis: 375 mg/m² einmal wöchentlich über 4 Wochen5).
Eine niedrige Dosis (100 mg/Woche × 4) wurde ebenfalls berichtet, um eine vollständige Remission zu erreichen2). Die vollständige Remissionsrate liegt bei 83–100 %2).
Therapieresistente/rezidivierende Fälle: Immunsuppressiva wie Vincristinsulfat und Cyclophosphamid sowie Splenektomie können in Betracht gezogen werden.
Ophthalmologische Behandlung der Netzhautischämie: Photokoagulation wird durchgeführt, um Neovaskularisationen zu behandeln.
QWarum ist eine Thrombozytentransfusion bei TTP kontraindiziert?
A
Bei TTP bilden sich thrombozytenreiche Mikrothromben in den Mikrogefäßen des gesamten Körpers. Eine Thrombozytentransfusion in diesem Zustand kann die Bildung weiterer Mikrothromben fördern und den Krankheitsverlauf verschlechtern. Die Erstlinientherapie ist der Plasmaaustausch, und Thrombozytentransfusionen sollten nur bei lebensbedrohlichen Blutungen durchgeführt werden.
QWie werden ophthalmologische Komplikationen behandelt?
A
Bei Netzhautischämie wird eine Photokoagulation durchgeführt. Sie dient der Behandlung von Neovaskularisationen, die als Folge der Netzhautischämie auftreten. Ein Papillenödem kann durch erhöhten intrakraniellen Druck oder maligne Hypertonie verursacht werden und muss parallel zur Behandlung der Grunderkrankung (TTP) gemanagt werden.
6. Pathophysiologie und detaillierter Pathomechanismus
Der Pathomechanismus der ophthalmologischen Zeichen bei TTP beruht auf einer Kaskade der Mikrothrombenbildung, die durch einen ADAMTS13-Mangel ausgelöst wird.
vWF ist ein Protein, das von Gefäßendothel und Megakaryozyten sezerniert wird und normalerweise durch ADAMTS13 gespalten und abgebaut wird. Bei erworbenem TTP bilden sich Autoantikörper gegen ADAMTS13, während bei angeborenem TTP Mutationen im ADAMTS13-Genlocus die Ursache sind.
Aufgrund eines Mangels oder einer verminderten Aktivität von ADAMTS13 akkumulieren im Blut hochmolekulare vWF-Multimere (übermolekulare vWF-Multimere), die resistent gegen abbauende Enzyme sind. Diese binden an Thrombozyten und bilden thrombozytenreiche Mikrothromben, die die kleinen Blutgefäße im ganzen Körper verstopfen. Eine Anämie durch mechanische Hämolyse (mikroangiopathische hämolytische Anämie) tritt gleichzeitig auf.
Mechanismus der Entwicklung ophthalmologischer Zeichen
Mikrothromben in den Netzhaut- und Aderhautgefäßen → Beeinträchtigung der Blutversorgung der Netzhaut → Netzhautblutungen, Gefäßverschlüsse und seröse Netzhautablösung treten auf.
Sekundäre Veränderungen durch Netzhautischämie → Bildung neuer Blutgefäße.
Hirnnervenstörungen infolge thrombotischer Ischämie → Anisokorie und Augenstellungsanomalien treten auf.
Die Prävalenz von Schlaganfällen bei TTP wird mit 13,9 % angegeben, was höher ist als die 6,3–7,8 % bei älteren Menschen im Allgemeinen 1). Das reversible posteriore Leukenzephalopathie-Syndrom (PRES) ist eine Erkrankung, die mit TTP einhergehen kann, und die diffusionsgewichtete MRT (DWI) ist nützlich, um zwischen vasogenem und zytotoxischem Ödem zu unterscheiden 1).
7. Aktuelle Forschung und zukünftige Perspektiven (Berichte aus der Forschungsphase)
Caplacizumab ist ein humanisierter Nanokörper (Einzeldomänen-Antikörper), der gegen die A1-Domäne des vWF gerichtet ist. In der HERCULES-Studie wurde eine schnellere Normalisierung der Thrombozytenzahl, eine Verringerung der Anzahl der Plasmaaustauschsitzungen und eine niedrigere Rückfallrate nachgewiesen 2)3). Auch die Wirksamkeit bei refraktärer TTP wurde berichtet 5).
In der Übersichtsarbeit von Xu et al (2024) zu TTP-Fällen in der Schwangerschaft wird gezeigt, dass rekombinantes ADAMTS13 möglicherweise inhibitorische Antikörper überwinden und die vWF-Spaltungsaktivität normalisieren kann, was es zu einer vielversprechenden zukünftigen Behandlungsoption macht3).
Galindo-Calvillo et al (2021) berichteten über einen Fall von rezidivierender TTP, der während der COVID-19-Pandemie ohne Plasmaaustausch allein mit niedrig dosiertem Rituximab (100 mg/Woche × 4) und Prednison (1 mg/kg) eine vollständige hämatologische Remission erreichte2). Dies deutet auf die Möglichkeit einer Behandlungsstrategie ohne Plasmaaustausch bei einer Untergruppe von Patienten mit stabiler rezidivierender TTP ohne Organbeteiligung hin.
Berichte über TTP nach BNT162b2-Impfung sind in der Literatur am häufigsten (7 von 10 Fällen), die meisten treten nach der zweiten Dosis auf5). Ein Epitop-Mimikry-Mechanismus wird vermutet, aber zur Bestätigung eines kausalen Zusammenhangs sind weitere Studien erforderlich.
Zhu H, Liu JY. Thrombotic thrombocytopenic purpura with neurological impairment: A Review. Medicine. 2022;101(49):e31851. doi:10.1097/MD.0000000000031851. PMID:36626546; PMCID:PMC9750575.
César David Galindo-Calvillo, Carlos Saúl Rodríguez-Roque, Andrés Gómez-De León, Luz Tarín-Arzaga, David Gómez-Almaguer. Treating thrombotic thrombocytopenic purpura without plasma exchange during the COVID-19 pandemic. A case report and a brief literature review. Transfusion and Apheresis Science. 2021;60(3):103107. doi:10.1016/j.transci.2021.103107.
Xu J, Tan LN, Li LX, Qiao GY. Case report of thrombotic thrombocytopenic purpura during pregnancy with a review of the relevant research. Medicine. 2024;103(20):e38112. doi:10.1097/MD.0000000000038112. PMID:38758904; PMCID:PMC11098172.
Shao X, Xu X, Li Q, Hu R, Tao K, Yang W, Dong A. Thrombotic Thrombocytopenia Purpura (TTP) following emergent aortic valve replacement after a complicated TAVR procedure: a case report and review of the literature. Journal of cardiothoracic surgery. 2024;19(1):545. doi:10.1186/s13019-024-03055-5. PMID:39313779; PMCID:PMC11418202.
Hammami E, Lamarque M, Aujoulat O, Debliquis A, Drénou B, Harzallah I. Acquired Thrombotic Thrombocytopenic Purpura After BNT162b2 COVID-19 Vaccine: Case Report and Literature Review. Laboratory medicine. 2022;53(6):e145-e148. doi:10.1093/labmed/lmac016. PMID:35482291; PMCID:PMC9129115.
Li P, Jiang J, Xi Q, Yang Z. An ADAMTS13 mutation that causes hereditary thrombotic thrombocytopenic purpura: a case report and literature review. BMC medical genomics. 2021;14(1):252. doi:10.1186/s12920-021-01099-3. PMID:34702267; PMCID:PMC8549186.
Lin HC, Huang J, Huang J, Zhang LJ, Yin XW, Yang JC, et al. Concurrence of immune thrombocytopenic purpura and thrombotic thrombocytopenic purpura: a case report and review of the literature. Journal of medical case reports. 2023;17(1):38. doi:10.1186/s13256-023-03762-y. PMID:36750960; PMCID:PMC9905008.
Wang Z, Xu H, Peng B, et al. Flavorubredoxin, a Candidate Trigger Related to Thrombotic Thrombocytopenic Purpura. Front Cell Infect Microbiol. 2022;12:864087.
Kopieren Sie den Artikeltext und fügen Sie ihn in den KI-Assistenten Ihrer Wahl ein.
Artikel in die Zwischenablage kopiert
Öffnen Sie unten einen KI-Assistenten und fügen Sie den kopierten Text in den Chat ein.