La púrpura trombótica trombocitopénica (PTT), también conocida como enfermedad de Moschcowitz, es una enfermedad sanguínea rara caracterizada por la pentada de anemia hemolítica microangiopática (AHMA), púrpura trombocitopénica, lesión renal aguda, anomalías neurológicas (alteración del estado mental) y fiebre.
Los signos oculares se reportan en el 14–20% de los casos, presentando diversos hallazgos debidos a la formación de microtrombos en los vasos retinianos y coroideos.
La incidencia se reporta de 3.7 a 11 casos por millón de personas al año. En la población adulta francesa, se reporta 1.5 casos por millón al año5). La prevalencia anual es de aproximadamente 10 casos por millón de personas7), y se sabe que es más frecuente en mujeres.
Diferencia de sexo: Las mujeres desarrollan la enfermedad de 2 a 3 veces más frecuentemente que los hombres6).
Antecedentes predisponentes: Más común en pacientes afrocaribeños y obesos.
Mortalidad sin tratamiento: Aproximadamente 90%. Con el intercambio de plasma, se reduce al 10–20%3).
La TTP se clasifica en tipos congénito y adquirido.
TTP congénito (síndrome de Upshaw-Schulman): Causado por mutaciones en el gen ADAMTS13. Representa aproximadamente el 5% de todos los casos. Se han identificado más de 260 sitios de mutación en la base de datos NCBI ClinVar, de los cuales aproximadamente el 60% son mutaciones sin sentido y aproximadamente el 20% son pequeñas deleciones o inserciones6).
TTP adquirido: Causado por autoanticuerpos contra ADAMTS13, representando aproximadamente el 95% de todos los casos3).
Q¿Con qué frecuencia ocurren los signos oculares de la TTP?
A
Los signos oculares se han reportado en el 14–20% de los casos. Incluyen hemorragias retinianas, oclusión vascular, desprendimiento seroso de retina y papiledema. Dado que los síntomas oculares pueden preceder a los síntomas sistémicos, el oftalmólogo puede ser el primero en sospechar TTP.
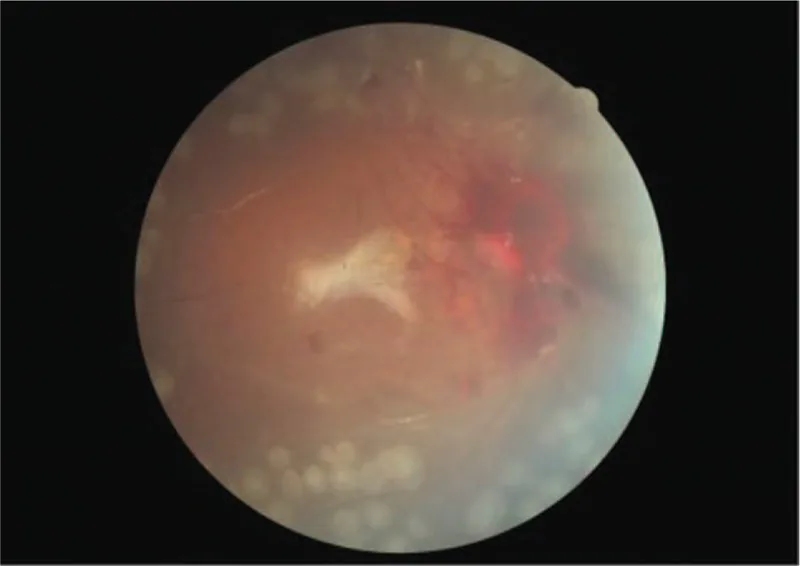
Bilateral proliferative retinopathy and ischemic optic neuropathy in a patient with atypical hemolytic-uremic syndrome: A case report. Medicine (Baltimore). 2019 Sep 27; 98(39):e17232. Figure 4. PMCID: PMC6775429. License: CC BY.
Fotografía en color del fondo de ojo del ojo derecho después de la cirugía. Aunque hay hemorragia visible alrededor del disco óptico, la retina está bien adherida.
Se han reportado los siguientes síntomas subjetivos oftalmológicos.
Pérdida rápida de la visión: Debido a lesiones retinianas.
Visión borrosa: Ocurre secundaria al edema de papila.
Diplopía: Asociada con trastornos del movimiento ocular por parálisis de nervios craneales.
Visión borrosa transitoria y visión amarilla: Reportado en casos de TTP asociada al embarazo3).
Los síntomas subjetivos sistémicos de la TTP incluyen cefalea, alteración de la conciencia, trastornos mentales, epilepsia, déficits neurológicos focales, fiebre, malestar general, artralgia, ictericia, náuseas y vómitos1). También se produce hemorragia cutáneo-mucosa (púrpura, petequias, hemorragia gingival) secundaria a trombocitopenia3).
La causa fundamental de la PTT es la disminución de la actividad de ADAMTS13. ADAMTS13 es una proteasa que escinde los multímeros grandes del factor de von Willebrand (vWF). Cuando su actividad se reduce, los multímeros grandes de vWF se acumulan en el torrente sanguíneo, formando microtrombos ricos en plaquetas.
Las mutaciones en el gen ADAMTS13 son la causa, y a menudo se manifiestan al ser desencadenadas por el embarazo. En la TTP congénita, el riesgo de recurrencia durante el embarazo alcanza el 100%, mientras que en la TTP adquirida se reporta entre 0 y 50%3).
Infección: Sepsis, infección por Salmonella, etc.8).
Embarazo: En el tercer trimestre, los niveles de vWF aumentan de 1.5 a 3 veces, y ADAMTS13 disminuye al 25-30% 3).
Enfermedades autoinmunes: LES, síndrome antifosfolípido (SAF), síndrome de Sjögren, etc.1)7).
Fármacos: Inhibidores de la tirosina quinasa, etc.
Vacunas: Se ha informado de aparición tras la vacunación contra la COVID-19, siendo más frecuente tras la vacuna BNT162b2 (7 de cada 10 casos en la literatura)5).
Cirugía: Existen casos reportados después de cirugía cardíaca (reemplazo valvular, TAVR)4).
Infección por VIH7).
Q¿La PTT es hereditaria?
A
La PTT congénita (síndrome de Upshaw-Schulman) es causada por mutaciones en el gen ADAMTS13 y es una enfermedad hereditaria. La base de datos NCBI ClinVar registra más de 260 sitios de mutación, a menudo en forma de mutaciones heterocigotas compuestas. Sin embargo, aproximadamente el 95% de todos los casos de PTT son adquiridos, causados por autoanticuerpos contra ADAMTS13.
El diagnóstico de TTP se basa principalmente en la confirmación de dos hallazgos: anemia hemolítica microangiopática (MAHA) y púrpura trombocitopénica. El tratamiento debe iniciarse sin esperar los resultados de la prueba de actividad de ADAMTS13.
En casos relacionados con el embarazo, es importante diferenciar de las siguientes enfermedades.
Síndrome HELLP, síndrome urémico hemolítico (SUH), hígado graso agudo del embarazo, SAF obstétrico3).
Otras enfermedades que presentan baja actividad de ADAMTS13, como sepsis, CID, enfermedad hepática y malaria por Plasmodium falciparum, también requieren diferenciación.
Si se encuentran hallazgos oculares, realice una anamnesis detallada de otros síntomas sistémicos y verifique la presencia de la pentada clásica de TTP.
El recambio plasmático es el tratamiento de primera línea. Se esperan los siguientes cuatro efectos.
Reposición de ADAMTS13
Eliminación de inhibidores (autoanticuerpos)
Eliminación de multímeros de vWF de peso molecular ultraalto (UL-vWFM)
Reposición de vWF normal
En la PTT asociada al embarazo, se realizan 2,000 mL/sesión (40–60 mL/kg) 1–2 veces al día 3). El recambio plasmático reduce la tasa de mortalidad no tratada del 90% al 10–20% 3).
Si no hay deficiencia grave adquirida de ADAMTS13, la infusión de plasma fresco congelado también es una opción.
Se utiliza como terapia adyuvante en casos refractarios o recurrentes.
Dosis estándar: 375 mg/m² una vez por semana durante 4 semanas5).
También se ha informado que dosis bajas (100 mg/semana × 4 dosis) logran remisión completa2). La tasa de remisión completa se reporta entre 83 y 100%2).
Casos refractarios/recurrentes: Se pueden considerar fármacos inmunosupresores como sulfato de vincristina y ciclofosfamida, y esplenectomía.
Tratamiento oftálmico para la isquemia retiniana: Se realiza fotocoagulación. Se lleva a cabo para tratar la neovascularización.
Q¿Por qué está contraindicada la transfusión de plaquetas en la TTP?
A
En la PTT, se forman microtrombos ricos en plaquetas en los microvasos de todo el cuerpo. La transfusión de plaquetas en este estado puede promover una mayor formación de microtrombos y empeorar la condición. El tratamiento de primera línea es el recambio plasmático, y la transfusión de plaquetas no debe realizarse excepto en casos de hemorragia que ponga en peligro la vida.
Q¿Cómo se realiza el tratamiento de las complicaciones oftálmicas?
A
Si se produce isquemia retiniana, se realiza fotocoagulación. Esto se hace para tratar la neovascularización secundaria a la isquemia retiniana. El papiledema puede deberse a hipertensión intracraneal o hipertensión maligna y debe manejarse en paralelo con el tratamiento de la enfermedad subyacente (PTT).
El mecanismo de los signos oftálmicos en la PTT se origina en una cascada de formación de microtrombos desencadenada por la deficiencia de ADAMTS13.
El vWF es una proteína secretada por las células endoteliales vasculares y los megacariocitos, y normalmente es escindido y degradado por ADAMTS13. En la PTT adquirida, se forman autoanticuerpos contra ADAMTS13, mientras que en la PTT congénita, las mutaciones en el locus del gen ADAMTS13 son la causa.
La deficiencia o disminución de la actividad de ADAMTS13 provoca la acumulación en el torrente sanguíneo de multímeros de vWF de alto peso molecular (multímeros de vWF de tamaño ultra grande) resistentes a las enzimas degradantes en el plasma. Estos se unen a las plaquetas formando microtrombos ricos en plaquetas que obstruyen los microvasos de todo el cuerpo. También se produce anemia por hemólisis mecánica (anemia hemolítica microangiopática).
Microtrombos en los vasos retinianos y coroideos → Alteración del suministro sanguíneo a la retina → Hemorragia retiniana, oclusión vascular y desprendimiento seroso de retina.
Cambios secundarios a la isquemia retiniana → Formación de nuevos vasos sanguíneos.
Trastornos de los nervios craneales secundarios a isquemia trombótica → Anisocoria y posición anormal del ojo.
La prevalencia de accidente cerebrovascular en la PTT se reporta en 13.9%, superior al 6.3–7.8% en la población general de edad avanzada 1). El síndrome de encefalopatía posterior reversible (PRES) es una condición que puede complicar la PTT, y la resonancia magnética con difusión (DWI) es útil para diferenciar entre edema vasogénico y edema citotóxico 1).
7. Investigación más reciente y perspectivas futuras (Informes en fase de investigación)
Caplacizumab es un nanocuerpo humanizado (anticuerpo de dominio único) dirigido al dominio A1 del vWF. En el ensayo HERCULES, se confirmó que acelera la normalización del recuento de plaquetas, reduce el número de sesiones de recambio plasmático y disminuye la tasa de recaídas2)3). También se ha informado su eficacia en TTP refractaria5).
En una revisión de casos de TTP asociada al embarazo por Xu et al (2024), se sugirió que el ADAMTS13 recombinante podría superar a los anticuerpos inhibidores y normalizar la actividad de escisión del vWF, por lo que se espera como una futura opción terapéutica3).
Galindo-Calvillo et al. (2021) reportaron un caso de TTP recidivante que logró remisión hematológica completa solo con rituximab en dosis bajas (100 mg/semana × 4 dosis) y prednisona (1 mg/kg) sin recambio plasmático durante la pandemia de COVID-192). Esto sugiere la posibilidad de una estrategia de tratamiento que no requiere recambio plasmático en un subgrupo de pacientes con TTP recidivante estable sin daño orgánico.
Los informes de TTP después de la vacunación con BNT162b2 son los más numerosos en la literatura (7 de 10 casos), y la mayoría ocurren después de la segunda dosis5). Se sospecha un mecanismo de mimetismo de epítopos, pero se necesita más investigación para establecer la causalidad.
Zhu H, Liu JY. Thrombotic thrombocytopenic purpura with neurological impairment: A Review. Medicine. 2022;101(49):e31851. doi:10.1097/MD.0000000000031851. PMID:36626546; PMCID:PMC9750575.
César David Galindo-Calvillo, Carlos Saúl Rodríguez-Roque, Andrés Gómez-De León, Luz Tarín-Arzaga, David Gómez-Almaguer. Treating thrombotic thrombocytopenic purpura without plasma exchange during the COVID-19 pandemic. A case report and a brief literature review. Transfusion and Apheresis Science. 2021;60(3):103107. doi:10.1016/j.transci.2021.103107.
Xu J, Tan LN, Li LX, Qiao GY. Case report of thrombotic thrombocytopenic purpura during pregnancy with a review of the relevant research. Medicine. 2024;103(20):e38112. doi:10.1097/MD.0000000000038112. PMID:38758904; PMCID:PMC11098172.
Shao X, Xu X, Li Q, Hu R, Tao K, Yang W, Dong A. Thrombotic Thrombocytopenia Purpura (TTP) following emergent aortic valve replacement after a complicated TAVR procedure: a case report and review of the literature. Journal of cardiothoracic surgery. 2024;19(1):545. doi:10.1186/s13019-024-03055-5. PMID:39313779; PMCID:PMC11418202.
Hammami E, Lamarque M, Aujoulat O, Debliquis A, Drénou B, Harzallah I. Acquired Thrombotic Thrombocytopenic Purpura After BNT162b2 COVID-19 Vaccine: Case Report and Literature Review. Laboratory medicine. 2022;53(6):e145-e148. doi:10.1093/labmed/lmac016. PMID:35482291; PMCID:PMC9129115.
Li P, Jiang J, Xi Q, Yang Z. An ADAMTS13 mutation that causes hereditary thrombotic thrombocytopenic purpura: a case report and literature review. BMC medical genomics. 2021;14(1):252. doi:10.1186/s12920-021-01099-3. PMID:34702267; PMCID:PMC8549186.
Lin HC, Huang J, Huang J, Zhang LJ, Yin XW, Yang JC, et al. Concurrence of immune thrombocytopenic purpura and thrombotic thrombocytopenic purpura: a case report and review of the literature. Journal of medical case reports. 2023;17(1):38. doi:10.1186/s13256-023-03762-y. PMID:36750960; PMCID:PMC9905008.
Wang Z, Xu H, Peng B, et al. Flavorubredoxin, a Candidate Trigger Related to Thrombotic Thrombocytopenic Purpura. Front Cell Infect Microbiol. 2022;12:864087.
Copia el texto del artículo y pégalo en el asistente de IA que prefieras.
Artículo copiado al portapapeles
Abre un asistente de IA abajo y pega el texto copiado en el chat.