Thrombotic thrombocytopenic purpura (TTP), also known as Moschcowitz disease, is a rare blood disorder characterized by the pentad of microangiopathic hemolytic anemia (MAHA), thrombocytopenic purpura, acute kidney injury, neurological abnormalities (altered mental status), and fever.
Ocular signs are reported in 14–20% of cases, presenting various findings due to microthrombi formation in retinal and choroidal vessels.
The incidence is reported to be 3.7 to 11 cases per million people per year. In the French adult population, it is reported as 1.5 cases per million per year5). The annual prevalence is approximately 10 cases per million people7), and it is known to occur more frequently in women.
Sex difference: Women develop the disease 2 to 3 times more often than men6).
Predisposing background: More common in Afro-Caribbean and obese patients.
Untreated mortality: Approximately 90%. With plasma exchange, it is reduced to 10–20%3).
TTP is classified into congenital and acquired types.
Congenital TTP (Upshaw-Schulman syndrome): Caused by ADAMTS13 gene mutations. Accounts for about 5% of all cases. Over 260 mutation sites have been identified in the NCBI ClinVar database, with approximately 60% being missense mutations and about 20% being small deletions or insertions6).
Acquired TTP: Caused by autoantibodies against ADAMTS13, accounting for about 95% of all cases3).
QHow often do ocular signs of TTP occur?
A
Ocular signs have been reported in 14–20% of cases. These include retinal hemorrhages, vascular occlusion, serous retinal detachment, and papilledema. Since ocular symptoms may precede systemic symptoms, ophthalmologists may be the first to suspect TTP.
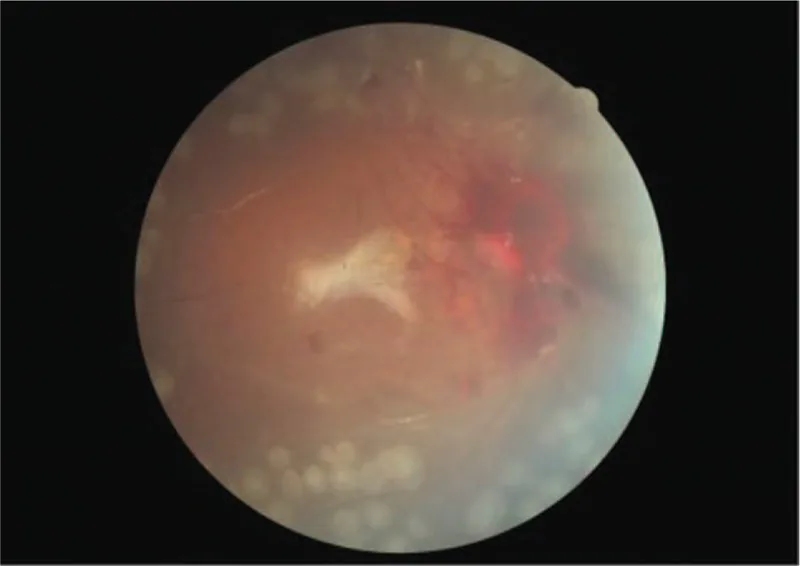
Bilateral proliferative retinopathy and ischemic optic neuropathy in a patient with atypical hemolytic-uremic syndrome: A case report. Medicine (Baltimore). 2019 Sep 27; 98(39):e17232. Figure 4. PMCID: PMC6775429. License: CC BY.
Fundus color photograph of the right eye after surgery. Although there is visible hemorrhage around the optic disc, the retina is well-attached.
The root cause of TTP is decreased ADAMTS13 activity. ADAMTS13 is a protease that cleaves von Willebrand factor (vWF) multimers. When its activity is reduced, large vWF multimers accumulate in the bloodstream, leading to the formation of platelet-rich microthrombi.
ADAMTS13 gene mutations are the cause, and they often become apparent when triggered by pregnancy. In congenital TTP, the risk of recurrence during pregnancy reaches 100%, while in acquired TTP it is reported to be 0–50%3).
Vaccines: Onset after COVID-19 vaccination has been reported, most frequently after BNT162b2 (7 out of 10 cases in the literature)5).
Surgery: Cases have been reported after cardiac surgery (valve replacement, TAVR)4).
HIV infection7).
QIs TTP inherited?
A
Congenital TTP (Upshaw-Schulman syndrome) is caused by mutations in the ADAMTS13 gene and is an inherited disorder. The NCBI ClinVar database lists over 260 mutation sites, often in the form of compound heterozygous mutations. However, about 95% of all TTP cases are acquired, caused by autoantibodies against ADAMTS13.
The diagnosis of TTP is primarily based on confirming two findings: microangiopathic hemolytic anemia (MAHA) and thrombocytopenic purpura. Treatment should be initiated without waiting for the results of ADAMTS13 activity testing.
Plasma exchange is the first-line treatment. The following four effects are expected.
Replenishment of ADAMTS13
Removal of inhibitors (autoantibodies)
Removal of ultra-large von Willebrand factor multimers (UL-vWFM)
Replenishment of normal vWF
In pregnancy-associated TTP, 2,000 mL/session (40–60 mL/kg) is performed 1–2 times per day 3). Plasma exchange reduces the untreated mortality rate from 90% to 10–20% 3).
If severe acquired ADAMTS13 deficiency is absent, infusion of fresh frozen plasma is also an option.
Refractory/recurrent cases: Immunosuppressive drugs such as vincristine sulfate and cyclophosphamide, and splenectomy may be considered.
Ophthalmic treatment for retinal ischemia: Photocoagulation is performed. This is done to address neovascularization.
QWhy is platelet transfusion contraindicated in TTP?
A
In TTP, platelet-rich microthrombi form in the microvasculature throughout the body. Platelet transfusion in this state may promote further microthrombus formation and worsen the condition. The first-line treatment is plasma exchange, and platelet transfusion should not be performed except in cases of life-threatening bleeding.
QHow is treatment for ophthalmic complications performed?
A
If retinal ischemia occurs, photocoagulation is performed. This is done to address neovascularization secondary to retinal ischemia. Papilledema may be due to increased intracranial pressure or malignant hypertension and must be managed in parallel with treatment of the underlying disease (TTP).
The mechanism of ophthalmic signs in TTP stems from a cascade of microthrombus formation triggered by ADAMTS13 deficiency.
vWF is a protein secreted by vascular endothelial cells and megakaryocytes, and is normally cleaved and degraded by ADAMTS13. In acquired TTP, autoantibodies against ADAMTS13 are formed, while in congenital TTP, mutations in the ADAMTS13 gene are the cause.
Deficiency or decreased activity of ADAMTS13 leads to accumulation of high-molecular-weight von Willebrand factor (ultra-large vWF multimers) resistant to cleavage in the plasma. These multimers bind to platelets, forming platelet-rich microthrombi that occlude small blood vessels throughout the body. Mechanical hemolysis causes anemia (microangiopathic hemolytic anemia).
Microthrombi in retinal and choroidal vessels → Impaired blood supply to the retina → Retinal hemorrhage, vascular occlusion, and serous retinal detachment.
Secondary changes due to retinal ischemia → Formation of new blood vessels.
Cranial nerve disorders secondary to thrombotic ischemia → Anisocoria and abnormal eye position.
The prevalence of stroke in TTP is reported to be 13.9%, which is higher than the 6.3–7.8% in the general elderly population 1). Posterior reversible encephalopathy syndrome (PRES) is a condition that can complicate TTP, and diffusion-weighted MRI (DWI) is useful for differentiating between vasogenic edema and cytotoxic edema 1).
7. Latest Research and Future Perspectives (Reports under Investigation)
Caplacizumab is a humanized nanobody (single-domain antibody) targeting the A1 domain of vWF. In the HERCULES trial, it was shown to accelerate platelet count normalization, reduce the number of plasma exchange sessions, and lower the relapse rate2)3). Its efficacy in refractory TTP has also been reported5).
In a review of pregnancy-associated TTP cases by Xu et al (2024), recombinant ADAMTS13 was suggested to potentially overcome inhibitory antibodies and normalize vWF cleavage activity, making it a promising future treatment option3).
Galindo-Calvillo et al (2021) reported a case of relapsed TTP that achieved complete hematologic remission with low-dose rituximab (100 mg/week × 4 doses) and prednisone (1 mg/kg) alone without plasma exchange during the COVID-19 pandemic2). This suggests the possibility of a treatment strategy that does not require plasma exchange in a subset of stable relapsed TTP patients without organ damage.
Reports of TTP onset after BNT162b2 vaccination are the most numerous in the literature (7 out of 10 cases), with most occurring after the second dose5). Epitope mimicry is suspected as the mechanism, but further research is needed to establish causality.
Zhu H, Liu JY. Thrombotic thrombocytopenic purpura with neurological impairment: A Review. Medicine. 2022;101(49):e31851. doi:10.1097/MD.0000000000031851. PMID:36626546; PMCID:PMC9750575.
César David Galindo-Calvillo, Carlos Saúl Rodríguez-Roque, Andrés Gómez-De León, Luz Tarín-Arzaga, David Gómez-Almaguer. Treating thrombotic thrombocytopenic purpura without plasma exchange during the COVID-19 pandemic. A case report and a brief literature review. Transfusion and Apheresis Science. 2021;60(3):103107. doi:10.1016/j.transci.2021.103107.
Xu J, Tan LN, Li LX, Qiao GY. Case report of thrombotic thrombocytopenic purpura during pregnancy with a review of the relevant research. Medicine. 2024;103(20):e38112. doi:10.1097/MD.0000000000038112. PMID:38758904; PMCID:PMC11098172.
Shao X, Xu X, Li Q, Hu R, Tao K, Yang W, Dong A. Thrombotic Thrombocytopenia Purpura (TTP) following emergent aortic valve replacement after a complicated TAVR procedure: a case report and review of the literature. Journal of cardiothoracic surgery. 2024;19(1):545. doi:10.1186/s13019-024-03055-5. PMID:39313779; PMCID:PMC11418202.
Hammami E, Lamarque M, Aujoulat O, Debliquis A, Drénou B, Harzallah I. Acquired Thrombotic Thrombocytopenic Purpura After BNT162b2 COVID-19 Vaccine: Case Report and Literature Review. Laboratory medicine. 2022;53(6):e145-e148. doi:10.1093/labmed/lmac016. PMID:35482291; PMCID:PMC9129115.
Li P, Jiang J, Xi Q, Yang Z. An ADAMTS13 mutation that causes hereditary thrombotic thrombocytopenic purpura: a case report and literature review. BMC medical genomics. 2021;14(1):252. doi:10.1186/s12920-021-01099-3. PMID:34702267; PMCID:PMC8549186.
Lin HC, Huang J, Huang J, Zhang LJ, Yin XW, Yang JC, et al. Concurrence of immune thrombocytopenic purpura and thrombotic thrombocytopenic purpura: a case report and review of the literature. Journal of medical case reports. 2023;17(1):38. doi:10.1186/s13256-023-03762-y. PMID:36750960; PMCID:PMC9905008.
Wang Z, Xu H, Peng B, et al. Flavorubredoxin, a Candidate Trigger Related to Thrombotic Thrombocytopenic Purpura. Front Cell Infect Microbiol. 2022;12:864087.
Copy the article text and paste it into your preferred AI assistant.
Article copied to clipboard
Open an AI assistant below and paste the copied text into the chat box.