Phakomatoses are a group of congenital disorders characterized by hamartomatous lesions in the skin, central nervous system, and eyes. They are also called neurocutaneous syndromes.
The term was coined by Dutch ophthalmologist Van der Hoeve, derived from the Greek word “phakos” (lens or spot). Initially, it included neurofibromatosis, tuberous sclerosis, and von Hippel-Lindau disease, later adding Sturge-Weber syndrome and ataxia telangiectasia. Currently, over 60 syndromes have been described.
The common pathological basis is an abnormality in the formation, migration, and differentiation of neural crest cells. Since neural crest cells are derived from the ectoderm and produce various cells such as Schwann cells and melanocytes, lesions occur in multiple organs including nerves, skin, and eyes. Known signaling pathways involved include RAS, MAPK/MEK, mTOR, PI3K/AKT, GNAQ, and VHL-HIF pathways.
The incidence of the six major diseases is as follows.
The six representative diseases are NF1, NF2, tuberous sclerosis, Sturge-Weber syndrome, von Hippel-Lindau disease, and ataxia telangiectasia. All are caused by genetic mutations and produce lesions in multiple organs including the nervous system, skin, and eyes. Currently, more than 60 syndromes are included in the category of phakomatoses.
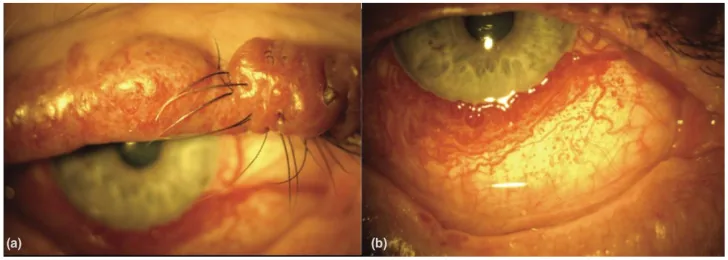
An Update on Multimodal Ophthalmological Imaging of Diffuse Choroidal Hemangioma in Sturge–Weber Syndrome. Vision (Basel). 2023 Oct 6; 7(4):64. Figure 1. PMCID: PMC10594527. License: CC BY.
(a) Port-wine stain of the upper lid with nodularity in a patient with Sturge–Weber syndrome. (b) Diffuse conjunctival vascularity in a patient with Sturge–Weber syndrome. From [15].
The types of ocular complications vary by disease, and subjective symptoms are also diverse.
NF1: As optic pathway tumors (optic gliomas) progress, visual acuity loss, color vision loss, and visual field defects occur. Plexiform neurofibromas may cause proptosis.
Tuberous sclerosis: Retinal astrocytic hamartomas are usually asymptomatic. Visual impairment occurs when the macula or optic disc is involved.
SWS: When associated with congenital glaucoma, corneal opacity, tearing, and photophobia occur. Late-onset glaucoma causes painless progressive visual field loss.
VHL: Asymptomatic in early stages. As retinal capillary hemangioma progresses, exudative changes, macular edema, and circinate retinopathy appear, leading to decreased vision.
Lisch nodules: The most common ocular finding in NF1. Light brown, well-defined, dome-shaped small nodules (<1–2 mm) appear multiple on the iris. Age-specific prevalence increases with age: <3 years 5%, 3–4 years 42%, 5–6 years 55%, ≥21 years 100%, and is included in the diagnostic criteria for NF1 (≥2 nodules). In Japanese individuals, the iris color is brown, so careful examination with a slit lamp is important.
Optic pathway tumors (optic gliomas): Occur in approximately 15–25% of NF1 patients. Most are low-grade pilocytic astrocytomas and are often asymptomatic. Progression can lead to optic atrophy, visual impairment, and visual field defects. Chiasmal involvement may occur.
Plexiform neurofibroma: Occurs in less than 10% of NF1 patients. Characterized by an “S-shaped deformity” of the eyelid, with a “bag of worms” texture on palpation. Can cause proptosis, strabismus, amblyopia, and congenital glaucoma.
Sphenoid wing dysplasia: Congenital defect of the orbital bone wall, which may cause pulsatile proptosis.
Glaucoma: Occurs in 1–2% of NF1 patients. There are two types: congenital (unilateral) and late-onset.
Exposure keratitis: Cranial nerve V and VII dysfunction due to bilateral acoustic neuromas causes facial numbness, diplopia, and lagophthalmos, leading to secondary exposure keratitis.
Lisch nodules are rare.
Ocular findings in tuberous sclerosis complex (TSC)
Retinal astrocytic hamartoma: Occurs in about 50% of cases. Classified into three types: (i) flat, translucent, noncalcified; (ii) elevated, multinodular, calcified (mulberry-like appearance); (iii) transitional type. Usually multiple lesions at the posterior pole.
Retinal depigmented lesions: Appear as “punched-out” lesions.
The three main features are (1) facial hemangioma in the trigeminal nerve distribution, (2) ipsilateral intracranial hemangioma, and (3) ipsilateral glaucoma or choroidal hemangioma.
Glaucoma: The most important ocular finding in SWS. Occurs in 30–70% of cases. Congenital glaucoma (birth to 4 years) accounts for about 60%, leading to buphthalmos, corneal opacity, and megalocornea. Etiology is thought to involve angle dysgenesis, elevated episcleral venous pressure, and choroidal hemangioma. It occurs frequently in the presence of eyelid hemangioma.
Choroidal hemangioma: Occurs in approximately 20–70% of cases. It is diffuse with indistinct borders, making identification difficult on routine fundus examination. The fundus appears “tomato ketchup-like.” It usually does not show a tendency to enlarge, but may cause exudative changes and exudative retinal detachment.
Others: Vascular dilation and tortuosity of the conjunctiva, episclera, and iris.
Ocular findings in VHL (von Hippel-Lindau disease)
Retinal capillary hemangioma (hemangioblastoma): Occurs in 43–85% of VHL patients (approximately 60% in Japanese reports). About one-third are bilateral and multiple. It commonly occurs in the temporal periphery of the retina and is observed as a reddish-orange nodule with dilated and tortuous feeding and draining vessels. The mean age of onset is 25 years, and it usually appears by age 30.
Progression leads to exudative changes, retinal hemorrhage, macular edema, circinate retinopathy, and decreased visual acuity.
Fluorescein angiography shows dye inflow into the afferent artery → efferent vein perfusion, with marked dye leakage from the tumor area in the early phase.
Bulbar conjunctival telangiectasia: The most common ocular finding. It usually develops by age 5–8 years and is seen in 80–90% of patients1).
Ocular motor abnormalities: Nystagmus, oculomotor apraxia, abnormal saccades, convergence and accommodation abnormalities, and strabismus are observed.
Visual acuity is usually preserved.
QHow do Lisch nodules change with age?
A
Lisch nodules associated with NF1 increase in frequency with age. They are present in only 5% of children under 3 years old, but are found in nearly 100% of individuals aged 21 and older. In young children, they may be difficult to detect even with slit-lamp examination, so age-adjusted interpretation is necessary.
The genetic characteristics of each disease are as follows.
NF1: Approximately 50% are de novo mutations. Nearly complete penetrance but variable expressivity. Neurofibromin functions as a RAS-GAP, converting GTP-RAS to GDP-RAS2).
NF2: Loss of function of the merlin protein. A tumor suppressor mainly expressed in Schwann cells and meningeal cells; its deficiency leads to schwannomas, meningiomas, and ependymomas.
TSC: Sporadic cases account for about two-thirds. TSC2 mutations account for 75–80% of sporadic cases and result in a more severe phenotype than TSC1 mutations1). Hamartin/tuberin directly inhibit the mTOR pathway2).
SWS: Sporadic, caused by somatic mosaic mutations in the GNAQ gene1). Port-wine birthmark (PWB) involving the entire first branch (V1) of the trigeminal nerve carries a high risk of ocular and neurological complications.
VHL: Penetrance is nearly 100%, with about 20% being de novo mutations. pVHL is involved in ubiquitination and proteasomal degradation of HIF-α2). Type 1 (without pheochromocytoma) accounts for about 80%, Type 2 (with pheochromocytoma) for about 20%2).
AT: Autosomal recessive inheritance; ATM kinase is involved in DNA double-strand break repair and genome stability maintenance 1). Risk of malignant tumors (lymphoid) and radiation sensitivity are markedly high.
The diagnostic criteria for NF1 (NIH criteria, Japanese Dermatological Association 2008) require meeting 2 or more of the following 7 items for a definitive diagnosis.
Six or more café-au-lait spots (prepubertal: ≥5 mm in diameter; postpubertal: ≥15 mm)
Two or more neurofibromas or one diffuse neurofibroma
Clinical criteria include bilateral vestibular schwannomas as a characteristic finding. Diagnosis may be more difficult than NF1 due to sparse skin symptoms. Genetic testing detects mutations in over 90% of cases. CT/MRI confirms bilateral acoustic neuromas.
A definitive diagnosis is made when two major features or one major feature plus two or more minor features are present. Identification of a pathogenic TSC1/TSC2 variant is recognized as an independent diagnostic criterion 1).
Fundus examination, fluorescein angiography, and OCT are used to evaluate retinal astrocytic hamartomas. The most important differential diagnosis is retinoblastoma. Differentiation is possible based on the absence of calcification in childhood, sparse feeding vessels, and associated systemic symptoms, but differentiation from mulberry tumors can be difficult.
According to the Roach diagnostic scale, the criteria require at least two of the following: facial port-wine stain, elevated intraocular pressure, and leptomeningeal angioma 1). Enhanced-depth OCT and MRI are useful for evaluating choroidal hemangiomas. Head CT shows calcification in the cerebral cortex.
Genetic testing can detect VHL mutations in nearly 100% of cases and is currently the main method for definitive diagnosis. Dilated fundus examination is recommended annually starting from age 1. Characteristic findings include reddish-orange spherical tumors in the peripheral retina with dilated and tortuous vessels. Fluorescein angiography is useful for detecting small peripheral hemangioblastomas, and OCT is used to evaluate small lesions.
For clinical diagnosis, the triad of ataxia, bulbar conjunctival telangiectasia, and abnormal eye movements is important. Laboratory findings include elevated serum AFP, elevated CA125, and ATM protein deficiency (Western blot). Bulbar conjunctival telangiectasia is a pathognomonic finding for this disease. MRI shows diffuse cerebellar atrophy in the posterior fossa, particularly of the vermis and hemispheres 1).
Lisch nodules: Asymptomatic hamartomas that do not require treatment.
Optic glioma: Surgical resection is considered for progressive cases, but visual function is lost and postoperative complications are frequent. Chemotherapy is indicated for cases with chiasmal infiltration.
Plexiform neurofibroma: Total surgical excision is difficult and recurrence is common. In progressive cases, orbital exenteration may be necessary. For unresectable cases, selumetinib (MEK inhibitor) is used as a novel treatment 1).
Glaucoma: Management follows standard glaucoma treatment.
For vestibular schwannoma, surgical resection is standard; for tumors less than 3 cm, hearing preservation is possible in 65% of cases 1). Exposure keratitis is managed with artificial tears, protective corneal glasses, and tarsorrhaphy if needed.
Ocular treatment for tuberous sclerosis complex (TSC)
Epilepsy: Vigabatrin is the first-line treatment for infantile partial seizures and infantile spasms1). SEGA (subependymal giant cell astrocytoma) is an indication for neurosurgical resection1).
Treatment for glaucoma differs depending on the age of onset.
Congenital glaucoma (early-onset type): Surgical treatment is essential. Trabeculotomy and goniotomy are selected. Careful attention to choroidal detachment and hemorrhage is required, and the risk of complications is higher than usual due to elevated episcleral venous pressure. There is a tendency for resistance to medical therapy.
Late-onset type: First, drug therapy is administered; if ineffective, surgery is considered.
Treatment of choroidal hemangioma is selected based on the presence or absence of exudative changes as follows.
Photocoagulation (first choice): Using an argon or dye laser, densely coagulate the retina around the hemangioma and the feeding vessels, then directly coagulate the tumor itself. For tumors larger than 2 disc diameters, coagulate the feeding artery and surrounding retina before directly coagulating the tumor.
Cryocoagulation: Performed transsclerally when photocoagulation is difficult due to peripheral fundus location or giant hemangioma.
Vitrectomy: Considered when exudative changes are severe, retinal detachment occurs, or proliferative changes develop.
There is no specific treatment for conjunctival telangiectasia. For immunodeficiency, prophylactic antibiotics and intravenous immunoglobulin (IVIg) are administered 1). Ataxia is managed with symptomatic treatment.
QAt what age should screening for retinal hemangioblastoma in VHL begin?
A
For VHL, annual dilated fundus examination is recommended from 1 year of age. It is important that all family members of patients with confirmed VHL gene mutations also undergo fundus examination, as early treatment can lead to a good visual prognosis.
The molecular mechanisms of each disease are described below.
NF1 and TSC (mTOR pathway)
NF1 (neurofibromin): Functions as a RAS-GAP. It promotes the conversion of GTP-bound RAS (active) to GDP-bound RAS (inactive). Mutation leads to persistent RAS activation, dysregulation of the MAP kinase pathway and PI3K-Akt-mTOR pathway, resulting in uncontrolled cell proliferation2).
TSC (hamartin/tuberin): A tumor suppressor complex that directly inhibits mTOR complexes 1 and 2. Mutation leads to mTOR overactivation, causing abnormalities in energy metabolism, protein/lipid synthesis, and cell survival2). TSC2 mutations result in a more severe phenotype than TSC1 mutations1).
VHL (HIF pathway)
pVHL: Component of the E3 ubiquitin ligase complex (elongin B/C, Cullin 2, RBX1). Under normal oxygen conditions, prolyl hydroxylase hydroxylates HIF-α → pVHL recognizes it → ubiquitination → proteasomal degradation.
VHL mutation: A constant pseudohypoxic state occurs → HIF-α accumulation → heterodimer formation with HIF-1β → transcriptional activation of VEGF, erythropoiesis, metabolism, and cell proliferation-related genes → vascular tumor formation2).
NF2 (merlin): Tumor suppressor protein expressed in Schwann cells and meningeal cells. Mutations lead to schwannomas, meningiomas, and ependymomas.
SWS (GNAQ somatic mosaic mutation): Abnormal G protein-coupled transmembrane signaling. The timing and location of the mutation in the embryo result in different expression patterns in the trigeminal nerve region, intracranial area, and eye1). GNAQ is also a common molecular basis for SWS, KTS, and PPV3).
AT (ATM kinase): Tumor suppressor involved in DNA double-strand break repair and genomic stability maintenance. Mutation → DNA repair defect → cancer predisposition, markedly increased radiosensitivity, and immunodeficiency1).
Chevalier et al. (2021) examined the association between phakomatoses and endocrine tumors and reported that the signaling pathways of NF1, TSC, and VHL (RAS-PI3K-Akt-mTOR pathway, HIF pathway) are commonly involved in the development of multiple endocrine neoplasms2).
QWhy do phakomatoses cause lesions in multiple organs?
A
The common pathophysiological basis of phakomatoses is abnormal formation, migration, and differentiation of neural crest cells. Neural crest cells are derived from the ectoderm and produce various cells such as Schwann cells and melanocytes, leading to lesions in multiple organs including the nervous system, skin, and eyes. In addition, mutations in common signaling pathways such as the RAS-mTOR pathway and VHL-HIF pathway promote tumor development across organs.
7. Latest Research and Future Perspectives (Research-stage Reports)
Clinical trials for NF2-related schwannomas and progressive tumors are being conducted by the INTUITT-NF2 consortium, with promising results reported 1).
A phase 2 trial targeting VHL-associated clear cell renal cell carcinoma and pancreatic neuroendocrine tumors is ongoing.
In the MK6482 trial by Chevalier et al. (2021), objective responses were observed in 64% of pancreatic neuroendocrine tumors, with a 12-month progression-free survival rate of 98.3% 2).
The EXIST trial for TSC-associated renal angiomyolipomas and SEGAs confirmed morphological responses, and application to endocrine tumors is also expected 2).
GNAQ gene mutations have been identified as a common molecular basis for SWS, Klippel-Trenaunay syndrome (KTS), and phakomatosis pigmentovascularis (PPV) 3). Due to the risk of choroidal melanoma in these overlapping cases, dilated fundus examinations 1–2 times per year are recommended 3).
Gama SM, Tamanini JVG, Moraes MPM, Silva TYT, Lima FT, Pedroso JL, et al. A diagnostic approach to neurocutaneous syndromes. Arquivos de neuro-psiquiatria. 2025;83(7):1-14. doi:10.1055/s-0045-1809664. PMID:40562378; PMCID:PMC12196566.
Chevalier B, Dupuis H, Jannin A, Lemaitre M, Do Cao C, Cardot-Bauters C, et al. Phakomatoses and Endocrine Gland Tumors: Noteworthy and (Not so) Rare Associations. Frontiers in endocrinology. 2021;12:678869. doi:10.3389/fendo.2021.678869. PMID:34025587; PMCID:PMC8134657.
Senthilkumar VA, Kohli P, Mishra C, et al. Ocular features in a patient presenting with a rare combination of multiple phakomatoses. BMJ Case Rep. 2022;15:e252746. doi:10.1136/bcr-2022-252746.
Copy the article text and paste it into your preferred AI assistant.
Article copied to clipboard
Open an AI assistant below and paste the copied text into the chat box.