母斑症(神經皮膚症候群)是一組先天性疾病的總稱,特徵是在皮膚、神經系統和眼睛中產生過誤瘤性病變。
代表性疾病包括NF1 、NF2、結節性硬化症、史德奇-韋伯症候群、馮·希佩爾-林道病和毛細血管擴張性運動失調症這六種疾病。
在NF1 中,超過90%的成人可見Lisch結節(虹膜 過誤瘤),這是診斷上重要的眼部表現。
在SWS中,30%至70%的患者發生青光眼 ,先天性青光眼 需要手術治療。
在VHL中,約60%的患者出現視網膜 毛細血管瘤 ,建議從1歲起每年進行一次散瞳 眼底檢查 。
每種疾病的眼部併發症常在無症狀時進展,因此定期的眼科管理至關重要。
分子標靶藥物(如MEK抑制劑、mTOR抑制劑)已被引入作為部分疾病的標準治療或新療法。
斑痣 性錯構瘤病是一組以皮膚、中樞神經系統和眼睛的錯構瘤性病變為特徵的先天性疾病總稱。也稱為神經皮膚症候群。
該命名由荷蘭眼科醫生Van der Hoeve提出,源自希臘語“phakos”(透鏡或斑點)。最初包括神經纖維瘤病 、結節性硬化症和馮·希佩爾-林道病,隨後加入了斯特奇-韋伯症候群和毛細血管擴張性運動失調症。目前已有超過60種症候群被描述。
共同的病理基礎是神經嵴細胞的形成、遷移和分化異常。由於神經嵴細胞來源於外胚層,產生許旺細胞、黑色素細胞等多種細胞,因此神經、皮膚和眼睛等多個器官會出現病變。已知涉及的訊號通路包括RAS、MAPK/MEK、mTOR、PI3K/AKT、GNAQ和VHL-HIF通路。
六大主要疾病的發生頻率如下。
疾病 發生率(每X人中1例) NF1 (神經纖維瘤病 1型)3,000~5,000 結節性硬化症(TSC ) 6,000~10,000 SWS(史德奇-韋伯症候群) 20,000~50,000 VHL(馮·希佩爾-林道病) 36,000 NF2(神經纖維瘤病 第2型) 25,000~100,000 毛細血管擴張性運動失調症(AT) 88,000至100,000以下
Q
母斑症包括哪些疾病?
A
代表性的6種疾病是NF1 、NF2、結節性硬化症、史德奇-韋伯症候群、馮·希佩爾-林道病和毛細血管擴張性運動失調症。所有這些疾病均由基因突變引起,並在神經、皮膚和眼等多器官產生病變。目前有60多種症候群被歸入母斑症的範疇。
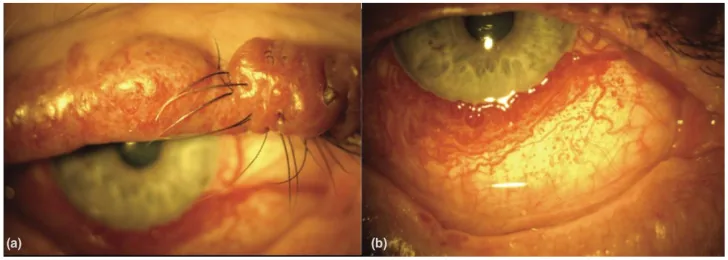
An Update on Multimodal Ophthalmological Imaging of Diffuse Choroidal Hemangioma in Sturge–Weber Syndrome. Vision (Basel). 2023 Oct 6; 7(4):64. Figure 1. PMCID: PMC10594527. License: CC BY.
(a) 斯特奇-韋伯症候群患者上眼瞼葡萄酒色斑伴結節。 (b) 斯特奇-韋伯症候群患者瀰漫性結膜 血管。摘自[15]。
各疾病的眼部併發症類型不同,自覺症狀也多樣。
NF1 視神經膠質瘤 )進展時,會出現視力 下降、色覺喪失和視野缺損 。叢狀神經纖維瘤可能導致眼球突出 。結節性硬化症 :視網膜 星形細胞錯構瘤通常無症狀。當累及黃斑 或視盤時,會導致視力 障礙。SWS :伴有先天性青光眼 時,會出現角膜 混濁、流淚和畏光 。遲發性青光眼 導致無痛性進行性視野缺損 。VHL :早期無症狀。視網膜微血管瘤 進展時出現滲出性變化、黃斑部水腫 、環狀白斑,導致視力 下降。AT :視力 通常維持正常。主要表現為眼球運動障礙 (眼震 、眼球運動失用)。
Lisch結節 :NF1 最常見的眼部表現。虹膜 上多發淡褐色、邊界清晰、圓頂狀小結節(<1–2 mm)。年齡別盛行率:<3歲5%,3–4歲42%,5–6歲55%,≥21歲100%,隨年齡增加,並納入NF1 診斷標準(≥2個)。日本人虹膜 顏色偏棕,因此裂隙燈 下仔細檢查很重要。視路腫瘤(視神經膠質瘤 ) :約15–25%的NF1 患者合併。多為低惡性度毛細胞星狀細胞瘤,常無症狀。進展時可導致視神經萎縮 、視力 障礙和視野缺損 。可能侵犯視交叉 。叢狀神經纖維瘤 :發生於不到10%的NF1 患者。特徵為眼瞼的「S形變形」,觸診時有「蟲袋」樣感覺。可導致眼球突出 、斜視 、弱視 和先天性青光眼 。蝶骨翼發育不全 :眼眶 骨壁的先天性缺損 ,可能引起搏動性眼球突出 。青光眼 NF1 患者。有先天性(單眼)和遲發性兩種型態。其他 :角膜 有髓神經纖維明顯、脈絡膜 增厚、視網膜有髓神經纖維 。
視神經鞘腦膜瘤 NF1 的視路膠質瘤形成對比的腫瘤類型。白內障 白內障 。視網膜 /RPE 過誤瘤視網膜 上膜暴露性角膜 炎 :雙側聽神經瘤導致第V、第VII腦神經功能障礙,引起面部麻木、複視 、兔眼 ,繼發暴露性角膜 炎。Lisch結節罕見。
視網膜 星形細胞過誤瘤視網膜 脫色素病變視網膜 血管異常玻璃體出血 、增殖性玻璃體視網膜病變 和視網膜剝離 。其他 :眼瞼血管纖維瘤、虹膜 脫色素斑、非典型脈絡膜 缺損 。
三大主要特徵為:(1)三叉神經 分布區的臉部血管瘤,(2)同側顱內血管瘤,(3)同側青光眼 或脈絡膜 血管瘤。
青光眼 先天性青光眼 (出生至4歲)約佔60%,導致牛眼、角膜 混濁和大角膜 。病因被認為涉及隅角 發育異常、鞏膜 上靜脈壓升高和脈絡膜 血管瘤。在眼瞼血管瘤存在時發生頻率高。脈絡膜 血管瘤眼底檢查 難以識別。眼底呈「番茄醬樣」外觀。通常無增大趨勢,但可引起滲出性變化和滲出性視網膜剝離 。其他 :結膜 、上鞏膜 和虹膜 的血管擴張迂曲。
視網膜 毛細血管瘤 (血管母細胞瘤)視網膜 顳側周邊部,表現為伴有擴張迂曲的輸入和輸出血管的橙紅色結節。平均發病年齡為25歲,通常在30歲前出現。進展可引起滲出性改變、視網膜 出血、黃斑 水腫、環狀白斑和視力 下降。
進一步進展可導致玻璃體出血 、牽拉性視網膜剝離 、新生血管性青光眼 和失明。
螢光眼底攝影 顯示染劑流入輸入動脈→輸出靜脈灌流,腫瘤區域早期出現明顯染劑滲漏。
球結膜 毛細血管擴張 :最常見的眼部表現。通常在5~8歲前出現,見於80%~90%的患者1) 。眼球運動障礙 眼震 、眼球運動失用、掃視異常、輻輳與調節異常、斜視 。視力 通常維持正常。
Q
Lisch結節隨年齡如何變化?
A
與NF1 相關的Lisch結節隨年齡增長出現頻率增加。3歲以下僅5%出現,但21歲以上幾乎100%可見。在年輕患者中,即使裂隙燈 檢查也可能難以檢測,因此需要考慮年齡因素進行解讀。
各疾病的遺傳形式、致病基因和染色體座位如下所示。
疾病 遺傳形式 致病基因 染色體座位 NF1 體染色體顯性 NF1 17q11.2 NF2 體染色體顯性 NF2 22q11.1-q13.1 TSC 體染色體顯性 TSC1/TSC2 9q34/16p13 VHL 體染色體顯性 VHL 3p25-26 SWS 散發性(體細胞鑲嵌) GNAQ 9q21 AT 體染色體隱性 ATM 11q22
各疾病的遺傳特徵如下。
NF1 2) 。NF2 :梅林蛋白功能喪失。主要表現於許旺細胞和腦膜細胞的腫瘤抑制因子,其缺失導致許旺瘤、腦膜瘤和室管膜瘤。TSC 1) 。錯構蛋白/結節蛋白直接抑制mTOR路徑2) 。SWS :散發性,由GNAQ基因體細胞嵌合突變引起1) 。累及三叉神經 第一支(V1)整個區域的葡萄酒色斑(PWB)具有較高的眼部和神經系統併發症風險。VHL :外顯率幾乎100%,約20%為新發突變。pVHL參與HIF-α的泛素化和蛋白酶體降解2) 。第1型(無嗜鉻細胞瘤)約佔80%,第2型(有嗜鉻細胞瘤)約佔20%2) 。AT :體染色體隱性遺傳 ,ATM激酶參與DNA雙股斷裂修復及基因組穩定性維持1) 。惡性腫瘤(淋巴系統)風險及放射線敏感性顯著增高。
NF1 的診斷標準(NIH標準、日本皮膚科學會2008年)需滿足以下7項中的2項或以上方可確診。
6個或更多咖啡牛奶斑(青春期前:最大直徑≥5mm;青春期後:≥15mm)
2個或更多神經纖維瘤或1個瀰漫性神經纖維瘤
腋窩或腹股溝區雀斑樣色素沉著
視神經膠質瘤 2個或更多Lisch虹膜 結節
特徵性骨病變(如蝶骨發育不良)
一等親患有相同疾病
眼科檢查的作用如下。
裂隙燈 顯微鏡眼前段OCT 和超音波生物顯微鏡 :有助於輔助檢測Lisch結節。MRI :視路腫瘤的最佳診斷方法。對視交叉 和視束的評估至關重要。視野檢查 光學同調斷層掃描 (OCT )視網膜神經纖維層 變薄。VEP (視覺誘發電位 )視神經 損傷。定期檢查間隔 :僅有Lisch結節→每年1次。伴有視神經膠質瘤 →每3個月1次。
臨床標準中,雙側前庭神經鞘瘤是特徵性表現。由於皮膚症狀不明顯,診斷可能比NF1 更困難。基因檢測可檢出90%以上的突變。CT/MRI可確認雙側聽神經瘤。
確診需符合2項主要特徵,或1項主要特徵加上2項以上次要特徵。TSC1/TSC2致病性變異的鑑定被認可為獨立的診斷標準1) 。
視網膜星狀細胞缺陷瘤 的評估使用眼底檢查 、螢光眼底攝影 和OCT 。最重要的鑑別診斷是視網膜母細胞瘤 。根據兒童期無鈣化、營養血管稀少、伴隨全身症狀的特點可進行鑑別,但與桑椹狀腫瘤的鑑別有時較困難。
Roach診斷量表要求至少符合面部葡萄酒色斑、眼壓 升高、軟腦膜血管瘤中的兩項1) 。增強深度OCT 和MRI對脈絡膜 血管瘤的評估有幫助。頭部CT顯示腦皮質內鈣化。
青光眼 的監測需要定期進行眼壓測量 、視神經 評估和視野檢查 。嬰幼兒可能需要在麻醉下進行裂隙燈 檢查、眼底檢查 和眼壓測量 。
基因檢測幾乎100%能檢測出VHL突變,是目前確診的主流方法。建議從1歲起每年進行一次散瞳 眼底檢查 。周邊部橙紅色球形腫瘤伴擴張迂曲血管是特徵性表現。螢光眼底造影有助於發現小的周邊部血管母細胞瘤,OCT 用於評估小病變。
臨床診斷中,共濟失調、球結膜 毛細血管擴張和眼球運動異常三聯徵很重要。實驗室檢查可見血清AFP升高、CA125升高和ATM蛋白缺失(Western blot)。球結膜 毛細血管擴張是本病的特徵性表現(pathognomonic)。MRI顯示後顱窩瀰漫性小腦萎縮,尤其是蚓部和半球1) 。
Lisch結節 :無症狀的錯構瘤,無需治療。視神經膠質瘤 視交叉 浸潤病例適用化學治療。蔓狀神經纖維瘤 :手術完全切除困難且易復發。進行病例可能需要眼眶 內容物剜除術。不可切除病例可使用司美替尼(MEK抑制劑)作為新療法1) 。青光眼 青光眼 治療進行管理。
前庭神經鞘瘤以手術切除為基本治療,小於3cm的腫瘤中65%可保留聽力1) 。暴露性角膜 炎使用人工淚液、角膜 保護眼鏡,必要時行瞼板 縫合處理。
視網膜 星形細胞過誤瘤視網膜 血管異常(動脈瘤樣擴張、動靜脈畸形)玻璃體出血 和視網膜剝離 的風險,需進行預防性光凝治療。若發生玻璃體出血 或視網膜剝離 ,考慮玻璃體 手術。癲癇 :嬰兒局部性發作和嬰兒點頭痙攣 的首選治療為氨己烯酸1) 。SEGA(室管膜下巨細胞星形細胞瘤)需神經外科手術切除1) 。
青光眼 的治療
先天性青光眼 (早發型)小樑切開術 或隅角 切開術。需注意脈絡膜 剝離和出血,由於鞏膜 上靜脈壓升高,併發症風險高於常規。對藥物治療往往有抗藥性。晚期發病型 :首先進行藥物治療,無效時考慮手術。
脈絡膜 血管瘤的治療
早期治療是原則。
光凝固(首選) :使用氬雷射或染料雷射,先緻密凝固血管瘤周圍的視網膜 和營養血管,然後直接凝固瘤體。對於大於2個視盤直徑的腫瘤,先凝固流入動脈和周圍視網膜 ,再直接凝固腫瘤。冷凍凝固 鞏膜 施行。玻璃體切除術 視網膜剝離 或出現增殖性變化時考慮。末期病例 :需要管理新生血管性青光眼 。如有家族史,應對所有家庭成員進行眼底檢查 ,早期治療可望獲得良好預後。
結膜 毛細血管擴張沒有特異性治療。對於免疫缺陷,進行預防性抗生素和靜脈注射免疫球蛋白(IVIg )治療1) 。共濟失調以對症治療為主。
Q
VHL視網膜血管瘤應從何時開始檢查?
A
對於VHL,建議從1歲開始每年進行一次散瞳 眼底檢查 。已確認VHL基因突變的患者的所有家庭成員也應接受眼底檢查 ,早期治療有望獲得良好的視功能預後。
各疾病的分子層級發病機制如下所示。
NF1與TSC(mTOR路徑)
NF1 (神經纖維蛋白)2) 。
TSC (錯構蛋白/結節蛋白)2) 。TSC2突變比TSC1突變導致更嚴重的表型1) 。
VHL(HIF路徑)
pVHL :E3泛素連接酶複合體(elongin B/C、Cullin 2、RBX1)的組成部分。在正常氧氣條件下,脯氨醯羥化酶將HIF-α羥化→pVHL辨識→泛素化→蛋白酶體降解。
VHL突變 :持續出現假性缺氧狀態→HIF-α累積→與HIF-1β形成異二聚體→VEGF、紅血球生成、代謝和細胞增殖相關基因的轉錄活化→血管腫瘤形成2) 。
NF2(merlin) :腫瘤抑制蛋白,在許旺細胞和軟腦膜細胞中表現。突變導致許旺瘤、腦膜瘤和室管膜瘤。SWS(GNAQ體細胞嵌合突變) :G蛋白相關跨膜訊息傳導異常。胚胎中突變的時間和位置導致三叉神經 區域、顱內和眼部的表現模式不同1) 。GNAQ也是SWS、KTS和PPV 的共同分子基礎3) 。AT(ATM激酶) :參與DNA雙股斷裂修復和基因組穩定性維持的腫瘤抑制因子。突變→DNA修復障礙→癌症易感性、放射敏感性顯著增加和免疫缺陷1) 。
Chevalier等人(2021)研究了母斑症與內分泌腫瘤的關聯,報告指出NF1 、TSC 和VHL的訊息傳導路徑(RAS-PI3K-Akt-mTOR路徑、HIF路徑)共同參與多發性內分泌腫瘤 的發生2) 。
Q
為什麼母斑症會在多個器官產生病變?
A
母斑症共同的病理生理基礎是神經嵴細胞的形成、遷移和分化異常。神經嵴細胞來源於外胚層,產生許旺細胞、黑色素細胞等多種細胞,因此病變會波及神經、皮膚、眼睛等多個器官。此外,RAS-mTOR路徑和VHL-HIF路徑等共同訊息傳導路徑的突變促進了跨器官的腫瘤發生。
一種獲得FDA核准用於不可切除的叢狀神經纖維瘤的分子標靶藥物。它靶向NF1 中MAPK路徑的過度活化1) 。
針對NF2相關許旺氏瘤和進展性腫瘤的臨床試驗由INTUITT-NF2聯盟進行,已報告有希望的結果1) 。
針對VHL相關透明細胞腎細胞癌和胰臟神經內分泌腫瘤的第二期試驗正在進行中。
Chevalier等人(2021)的MK6482試驗中,64%的胰臟神經內分泌腫瘤達到客觀緩解,12個月無惡化存活率為98.3% 2) 。
針對TSC 相關腎血管平滑肌脂肪瘤和SEGA的EXIST試驗證實了形態學緩解,其在內分泌腫瘤中的應用也備受期待 2) 。
GNAQ基因突變已被確定為SWS、克利佩爾-特雷諾內症候群(KTS)和色素血管性母斑症(PPV )的共同分子基礎 3) 。這些重疊病例有脈絡膜黑色素瘤 的風險,因此建議每年進行1-2次散瞳 眼底檢查 3) 。
Gama SM, Tamanini JVG, Moraes MPM, Silva TYT, Lima FT, Pedroso JL, et al. A diagnostic approach to neurocutaneous syndromes. Arquivos de neuro-psiquiatria. 2025;83(7):1-14. doi:10.1055/s-0045-1809664. PMID:40562378; PMCID:PMC12196566.
Chevalier B, Dupuis H, Jannin A, Lemaitre M, Do Cao C, Cardot-Bauters C, et al. Phakomatoses and Endocrine Gland Tumors: Noteworthy and (Not so) Rare Associations. Frontiers in endocrinology. 2021;12:678869. doi:10.3389/fendo.2021.678869. PMID:34025587; PMCID:PMC8134657.
Senthilkumar VA, Kohli P, Mishra C, et al. Ocular features in a patient presenting with a rare combination of multiple phakomatoses. BMJ Case Rep. 2022;15:e252746. doi:10.1136/bcr-2022-252746.
開啟下方的 AI 助手,並將複製的內容貼到聊天欄。