فاکوماتوزها (phakomatoses) گروهی از بیماریهای مادرزادی هستند که با ضایعات هامارتومایی در پوست، سیستم عصبی مرکزی و چشم مشخص میشوند. به این بیماریها سندرمهای عصبی-پوستی (neurocutaneous syndromes) نیز گفته میشود.
نامگذاری این بیماریها توسط چشمپزشک هلندی، ون در هووه (Van der Hoeve)، انجام شده و از کلمه یونانی «phakos» (به معنی عدس یا لکه) گرفته شده است. در ابتدا سه بیماری نوروفیبروماتوز، توبروز اسکلروزیس و بیماری فون هیپل-لیندو را شامل میشد و بعدها سندرم استورج-وبر و آتاکسی تلانژکتازی به آن اضافه شد. امروزه بیش از 60 سندرم توصیف شده است.
زمینه پاتولوژیک مشترک این بیماریها، ناهنجاری در تشکیل، مهاجرت و تمایز سلولهای تاج عصبی است. سلولهای تاج عصبی از اکتودرم منشأ گرفته و سلولهای شوان، ملانوسیت و سایر سلولها را تولید میکنند؛ بنابراین ضایعات در اندامهای متعدد عصبی، پوستی و چشمی ایجاد میشود. مسیرهای سیگنالینگ درگیر شامل RAS، MAPK/MEK، mTOR، PI3K/AKT، GNAQ و VHL-HIF هستند.
شش بیماری اصلی عبارتند از NF1، NF2، توبروز اسکلروزیس، سندرم استورج-وبر، بیماری فون هیپل-لیندو و آتاکسی تلانژکتازی. همه این بیماریها ناشی از جهشهای ژنتیکی هستند و ضایعاتی در سیستم عصبی، پوست و چشم ایجاد میکنند. در حال حاضر بیش از 60 سندرم در دسته فاکوماتوزها قرار میگیرند.
An Update on Multimodal Ophthalmological Imaging of Diffuse Choroidal Hemangioma in Sturge–Weber Syndrome. Vision (Basel). 2023 Oct 6; 7(4):64. Figure 1. PMCID: PMC10594527. License: CC BY.
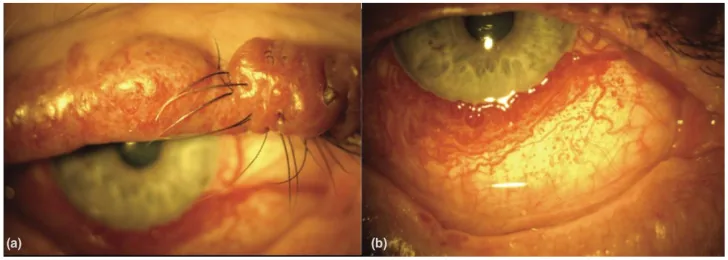
(a) لکه شرابی پلک فوقانی با ندولاریته در بیمار مبتلا به سندرم استورج-وبر. (b) عروق منتشر ملتحمه در بیمار مبتلا به سندرم استورج-وبر. از [15].
نوع عوارض چشمی در هر بیماری متفاوت است و علائم ذهنی نیز متنوع هستند.
NF1: با پیشرفت تومور مسیر بینایی (گلیوم عصب بینایی)، کاهش بینایی، از دست دادن دید رنگی و نقص میدان بینایی رخ میدهد. نوروفیبروم پلکسیفرم ممکن است باعث بیرونزدگی کره چشم شود.
اسکلروز توبروز: هامارتوم سلولهای ستارهای شبکیه معمولاً بدون علامت است. در صورت درگیری ماکولا یا دیسک بینایی، اختلال بینایی ایجاد میشود.
SWS: در صورت همراهی با گلوکوم مادرزادی، کدورت قرنیه، اشکریزش و فوتوفوبیا رخ میدهد. در گلوکوم دیررس، نقص میدان بینایی پیشرونده بدون درد ایجاد میشود.
VHL: در مراحل اولیه بدون علامت است. با پیشرفت همانژیوم مویرگی شبکیه، تغییرات اگزوداتیو، ادم ماکولا و رتینوپاتی حلقوی سفید ظاهر شده و منجر به کاهش بینایی میشود.
ندولهای لیش (Lisch nodules): شایعترین یافته چشمی در NF1. ندولهای کوچک (کمتر از 1-2 میلیمتر) به رنگ قهوهای روشن، با مرز مشخص و گنبدی شکل که به صورت متعدد در عنبیه ظاهر میشوند. شیوع بر اساس سن: زیر 3 سال 5%، 3-4 سال 42%، 5-6 سال 55%، بالای 21 سال 100% و با افزایش سن بیشتر میشود. در معیارهای تشخیصی NF1 (وجود 2 یا بیشتر) گنجانده شده است. در ژاپنیها به دلیل رنگ قهوهای عنبیه، معاینه با لامپ اسلیت ضروری است.
تومورهای مسیر بینایی (گلیوم عصب بینایی): در حدود 15-25% از بیماران NF1 رخ میدهد. اغلب آستروسیتوم پیلوسیتیک با درجه پایین است و معمولاً بدون علامت. با پیشرفت، باعث آتروفی عصب بینایی، اختلال بینایی و نقص میدان بینایی میشود. مواردی از درگیری کیاسم نیز وجود دارد.
نوروفیبروم پلکسیفرم: در کمتر از ۱۰٪ از بیماران NF1 رخ میدهد. با تغییر شکل «S شکل» پلک مشخص میشود و در لمس، احساس «کیسه کرم» دارد. میتواند باعث برونچشمی، استرابیسم، آمبلیوپی و گلوکوم مادرزادی شود.
دیسپلازی بال بزرگ استخوان اسفنوئید: نقص مادرزادی دیواره استخوانی کاسه چشم که میتواند باعث برونچشمی ضرباندار شود.
گلوکوم: در ۱ تا ۲٪ از موارد NF1 رخ میدهد. دو نوع مادرزادی (یکطرفه) و دیررس وجود دارد.
سایر موارد: برجسته شدن اعصاب میلیندار قرنیه، ضخیم شدن مشیمیه، اعصاب میلیندار شبکیه.
همارتریوم سلول ستارهای شبکیه: در حدود 50% موارد همراه است. به سه نوع تقسیم میشود: (i) نوع مسطح، نیمهشفاف و غیرکلسیفیه، (ii) نوع برجسته، چندندوله و کلسیفیه (ظاهر توتمانند)، (iii) نوع انتقالی. معمولاً چندین عدد در قطب خلفی دیده میشود.
ضایعات دپیگمانته شبکیه: ظاهری “پانچشده” (punched-out) دارند.
ناهنجاریهای عروق شبکیه: گشادشدگی آنوریسمی و ناهنجاری شریانی-وریدی ایجاد میکنند و میتوانند باعث خونریزی زجاجیه، رتینوپاتی پرولیفراتیو و جداشدگی شبکیه شوند.
سایر موارد: آنژیوفیبروم پلک، لکههای دپیگمانته عنبیه، و کلوبوم غیرمعمول مشیمیه.
سه علامت اصلی عبارتند از: (1) همانژیوم صورت در ناحیه عصب سهقلو، (2) همانژیوم داخل جمجمهای همطرف، و (3) گلوکوم یا همانژیوم مشیمیه همطرف.
گلوکوم: مهمترین یافته چشمی در SWS. در ۳۰ تا ۷۰٪ موارد رخ میدهد. گلوکوم مادرزادی (از بدو تولد تا ۴ سالگی) حدود ۶۰٪ موارد را تشکیل میدهد و باعث بوفتالموس، کدورت قرنیه و مگالوکورنه میشود. علت آن ناهنجاری زاویه، افزایش فشار ورید اپیاسکلرا و نقش همانژیوم مشیمیه در نظر گرفته میشود. در حضور همانژیوم پلک با فراوانی بیشتری رخ میدهد.
همانژیوم کوروئید: در حدود ۲۰ تا ۷۰٪ موارد رخ میدهد. به دلیل منتشر بودن و مرز نامشخص، شناسایی آن در معاینه معمول فوندوس دشوار است. ظاهر فوندوس شبیه «سس گوجهفرنگی» است. معمولاً تمایل به رشد ندارد، اما ممکن است باعث تغییرات اگزوداتیو و جداشدگی اگزوداتیو شبکیه شود.
سایر موارد: گشاد شدن و پیچخوردگی عروق ملتحمه، اپیاسکلرا و عنبیه.
همانژیوم مویرگی شبکیه (همانژیوبلاستوم): در ۴۳ تا ۸۵٪ بیماران VHL (حدود ۶۰٪ در گزارشهای داخلی) رخ میدهد. حدود یکسوم موارد دوطرفه و چندگانه هستند. در ناحیه تمپورال محیطی شبکیه شایعتر است و به صورت ندول قرمز-نارنجی با عروق آوران و وابران گشاد و پیچخورده مشاهده میشود. میانگین سن شروع ۲۵ سال است و معمولاً تا ۳۰ سالگی ظاهر میشود.
با پیشرفت، تغییرات اگزوداتیو، خونریزی شبکیه، ادم ماکولا، اگزودای حلقوی و کاهش بینایی رخ میدهد.
تلانژکتازی ملتحمه بولبار: شایعترین یافته چشمی. معمولاً تا سن ۵ تا ۸ سالگی ایجاد میشود و در ۸۰ تا ۹۰٪ بیماران دیده میشود1).
اختلالات حرکات چشم: نیستاگموس، آپراکسی حرکات چشم (oculomotor apraxia)، ناهنجاری ساکاد، ناهنجاری همگرایی و تطابق، و استرابیسم مشاهده میشود.
بینایی معمولاً حفظ میشود.
Qندولهای لیش (Lisch nodules) با افزایش سن چگونه تغییر میکنند؟
A
ندولهای لیش همراه با NF1 با افزایش سن بیشتر ظاهر میشوند. در کودکان زیر 3 سال تنها در 5% موارد دیده میشود، اما در افراد بالای 21 سال تقریباً در 100% موارد یافت میشود. در سنین پایین ممکن است حتی با معاینه با لامپ شکاف نیز تشخیص آن دشوار باشد و تفسیر باید با در نظر گرفتن سن انجام شود.
NF1: حدود 50% جهشهای de novo دارند. نفوذ تقریباً کامل است اما فنوتیپ متنوع است. نوروفیبرومین به عنوان RAS-GAP عمل میکند و GTP-RAS را به GDP-RAS تبدیل میکند2).
NF2: از دست دادن عملکرد پروتئین مرلین. یک مهارکننده تومور است که عمدتاً در سلولهای شوان و سلولهای لپتومننژ بیان میشود و کمبود آن باعث ایجاد شوانوما، مننژیوما و اپاندیموما میشود.
TSC: موارد پراکنده حدود دو سوم را تشکیل میدهند. جهش TSC2 75-80% موارد پراکنده را تشکیل میدهد و فنوتیپ شدیدتری نسبت به جهش TSC1 دارد1). هامارتین/توبرین مستقیماً مسیر mTOR را مهار میکند2).
SWS: پراکنده و ناشی از جهش موزاییک سوماتیک در ژن GNAQ است1). خال شرابی (PWB) در کل ناحیه اولین شاخه عصب سهقلو (V1) خطر عوارض چشمی و عصبی را افزایش میدهد.
VHL: نفوذ تقریباً 100% است و حدود 20% جهشهای de novo دارند. pVHL در یوبیکوئیتینه کردن و تجزیه پروتئازومی HIF-α نقش دارد2). نوع 1 (بدون فئوکروموسیتوما) حدود 80% و نوع 2 (با فئوکروموسیتوما) حدود 20% است2).
AT: یک بیماری اتوزومال مغلوب که در آن کیناز ATM در ترمیم شکستهای دو رشتهای DNA و حفظ پایداری ژنوم نقش دارد 1). خطر تومورهای بدخیم (لنفاوی) و حساسیت به پرتو به طور قابل توجهی بالا است.
در معیارهای بالینی، شوانوم دوطرفه عصب دهلیزی یافته مشخصه است. به دلیل کمبود علائم پوستی، تشخیص ممکن است دشوارتر از NF1 باشد. در آزمایش ژنتیک، بیش از ۹۰٪ جهشها شناسایی میشوند. تومورهای دوطرفه عصب شنوایی با CT و MRI تأیید میشوند.
تشخیص قطعی با دو معیار اصلی یا یک معیار اصلی به همراه دو یا چند معیار فرعی داده میشود. شناسایی جهشهای بیماریزای TSC1/TSC2 به عنوان یک معیار تشخیصی مستقل پذیرفته شده است 1).
برای ارزیابی هامارتوم سلولهای ستارهای شبکیه از معاینه فوندوس، آنژیوگرافی فلورسئین و OCT استفاده میشود. مهمترین تشخیص افتراقی رتینوبلاستوما است. در دوران کودکی، عدم وجود کلسیفیکاسیون، عروق تغذیهکننده کم و وجود علائم سیستمیک امکان افتراق را فراهم میکند، اما افتراق از تومور توتتوت شکل ممکن است دشوار باشد.
در مقیاس تشخیصی Roach، وجود حداقل دو مورد از سه علامت خال شرابی صورت، افزایش فشار چشم و آنژیوم لپتومننژال به عنوان معیار در نظر گرفته میشود 1). برای ارزیابی آنژیوم کوروئید، ED-OCT و MRI مفید هستند. در CT سر، کلسیفیکاسیون داخل قشر مغز مشاهده میشود.
برای پایش گلوکوم، اندازهگیری فشار چشم، ارزیابی عصب بینایی و آزمایش میدان بینایی به طور منظم انجام میشود. در نوزادان، علاوه بر معاینه با لامپ شکاف، ممکن است معاینه فوندوس و اندازهگیری فشار چشم تحت بیهوشی لازم باشد.
آزمایش ژنتیکی تقریباً در ۱۰۰٪ موارد جهش VHL را تشخیص میدهد و در حال حاضر روش اصلی تشخیص قطعی است. معاینه چشم با مردمک گشاد شده از سن ۱ سالگی سالی یک بار توصیه میشود. تومورهای کروی نارنجی-قرمز محیطی همراه با عروق مارپیچی و گشاد شده یافتههای مشخصه هستند. آنژیوگرافی فلورسئینی برای تشخیص همانژیوبلاستومهای کوچک محیطی مفید است و OCT برای ارزیابی ضایعات کوچک استفاده میشود.
در تشخیص بالینی، سهگانه آتاکسی، تلانژکتازی ملتحمهای و ناهنجاری حرکات چشم مهم است. افزایش AFP سرم، افزایش CA125 و کمبود پروتئین ATM (وسترن بلات) به عنوان یافتههای آزمایشگاهی دیده میشود. تلانژکتازی ملتحمهای یک یافته اختصاصی (پاتوگنومونیک) برای این بیماری است. MRI آتروفی منتشر مخچه در حفره خلفی (به ویژه ورمیس و نیمکرهها) را نشان میدهد1).
ندولهای لیش: هامارتوم بدون علامت هستند و نیازی به درمان ندارند.
گلیوم عصب بینایی: در موارد پیشرونده، برداشتن جراحی در نظر گرفته میشود، اما عملکرد بینایی از دست میرود و عوارض بعد از عمل زیاد است. در موارد نفوذ به کیاسم، شیمیدرمانی اندیکاسیون دارد.
نوروفیبروم پلکسیفرم: برداشتن کامل جراحی دشوار است و عود شایع است. در موارد پیشرونده، ممکن است تخلیه محتویات حدقه لازم شود. در موارد غیرقابل برداشت، سلومتینیب (مهارکننده MEK) به عنوان درمان جدید استفاده میشود1).
گلوکوم: مدیریت مطابق با درمان استاندارد گلوکوم انجام میشود.
برای شوانوم دهلیزی، برداشتن جراحی اساس درمان است و در تومورهای کوچکتر از 3 سانتیمتر، حفظ شنوایی در 65% موارد امکانپذیر است1). کراتیت اکسپوژر با اشک مصنوعی، عینک محافظ قرنیه و در صورت نیاز تارسورافی درمان میشود.
همارتوما سلولهای ستارهای شبکیه: معمولاً تمایل به رشد ندارد و نیازی به درمان نیست.
ناهنجاریهای عروقی شبکیه (اتساع آنوریسمی و ناهنجاری شریانی-وریدی): به دلیل خطر خونریزی زجاجیه و جداشدگی شبکیه، فتوکواگولاسیون پیشگیرانه انجام میشود. در صورت بروز خونریزی زجاجیه یا جداشدگی شبکیه، ویترکتومی در نظر گرفته میشود.
صرع: برای تشنجهای جزئی نوزادی و اسپاسمهای نوزادی، ویگاباترین درمان خط اول است1). SEGA (آستروسیتوم سلول غولپیکر ساباپاندیمال) نشانهای برای برداشتن جراحی مغز و اعصاب است1).
گلوکوم مادرزادی (شروع زودرس): درمان جراحی ضروری است. ترابکولوتومی و گونیوتومی انتخاب میشوند. باید به جداشدگی مشیمیه و خونریزی توجه کافی شود و به دلیل افزایش فشار ورید اپیاسکلرا، خطر عوارض بیشتر از حد معمول است. مقاومت به درمان دارویی معمولاً زیاد است.
نوع دیررس: ابتدا درمان دارویی انجام میشود و در صورت عدم پاسخ، جراحی مد نظر قرار میگیرد.
درمان همانژیوم کوروئید بر اساس وجود یا عدم وجود تغییرات اگزوداتیو از موارد زیر انتخاب میشود.
در صورت بروز تغییرات اگزوداتیو → فتوکوآگولاسیون
عدم پاسخ به فتوکوآگولاسیون یا جداشدگی اگزوداتیو شبکیه → پرتودرمانی (دوز کل حدود ۲۰ گری). انتظار میرود که جداشدگی تاولی شبکیه جا بیفتد و همانژیوم کوچک شود.
فتوکواگولاسیون (خط اول): با استفاده از لیزر آرگون یا دای، ابتدا شبکیه اطراف آنژیوم و عروق تغذیهکننده به طور متراکم منعقد میشوند، سپس خود تومور مستقیماً منعقد میشود. برای تومورهای بزرگتر از ۲ قطر دیسک بینایی، ابتدا شریان ورودی و شبکیه اطراف منعقد شده و سپس تومور مستقیماً منعقد میشود.
کرایوکواگولاسیون: در مواردی که فتوکواگولاسیون برای آنژیومهای محیطی یا بزرگ مشکل است، به صورت ترانس اسکلرال انجام میشود.
ویترکتومی: در موارد تغییرات اگزوداتیو شدید، جداشدگی شبکیه، یا تغییرات پرولیفراتیو در نظر گرفته میشود.
مراحل پایانی: نیاز به مدیریت گلوکوم نئوواسکولار وجود دارد.
در صورت وجود سابقه خانوادگی، معاینه فوندوس برای همه اعضای خانواده انجام شود و درمان زودهنگام میتواند پیشآگهی خوبی داشته باشد.
هیچ درمان اختصاصی برای تلانژکتازی ملتحمه وجود ندارد. برای نقص ایمنی، آنتیبیوتیکهای پیشگیرانه و ایمونوگلوبولین داخل وریدی (IVIg) تجویز میشود1). درمان آتاکسی عمدتاً علامتی است.
Qچه زمانی باید از نظر همانژیوم شبکیه در VHL غربالگری شود؟
A
در VHL، معاینه سالانه چشم با گشاد کردن مردمک از سن ۱ سالگی توصیه میشود. همچنین تمام اعضای خانواده بیمارانی که جهش ژن VHL در آنها تأیید شده است باید معاینه چشم انجام دهند، زیرا درمان زودهنگام میتواند پیشآگهی خوبی برای عملکرد بینایی داشته باشد.
مکانیسمهای مولکولی بروز هر بیماری در زیر آورده شده است.
NF1 و TSC (مسیر mTOR)
NF1 (نوروفیبرومین): به عنوان RAS-GAP عمل میکند. تبدیل RAS متصل به GTP (فعال) به RAS متصل به GDP (غیرفعال) را تسریع میکند. جهش → فعالسازی مداوم RAS → عدم تنظیم مسیر MAP کیناز و مسیر PI3K-Akt-mTOR → تکثیر سلولی کنترلنشده 2).
TSC (هامارتین/توبرین): یک کمپلکس سرکوبگر تومور است که مستقیماً mTOR کمپلکس ۱ و ۲ را مهار میکند. جهش → بیشفعالی mTOR → ناهنجاری در متابولیسم انرژی، سنتز پروتئین/لیپید و بقای سلولی 2). جهش TSC2 نسبت به جهش TSC1 منجر به فنوتیپ شدیدتری میشود 1).
VHL (مسیر HIF)
pVHL: جزء کمپلکس E3 یوبیکوئیتین لیگاز (الونگین B/C، کولین 2، RBX1). در شرایط نرمال اکسیژن، پرولیل هیدروکسیلاز HIF-α را هیدروکسیله میکند → pVHL آن را تشخیص میدهد → یوبیکوئیتینه میشود → توسط پروتئازوم تجزیه میشود.
جهش VHL: حالت شبه هیپوکسی به طور مزمن رخ میدهد → تجمع HIF-α → تشکیل هترودایمر با HIF-1β → فعالسازی رونویسی ژنهای مرتبط با VEGF، تولید گلبول قرمز، متابولیسم و تکثیر سلولی → تشکیل تومورهای عروقی2).
NF2 (مرلین): پروتئین سرکوبگر تومور که در سلولهای شوان و سلولهای لپتومننژ بیان میشود. جهش آن باعث ایجاد شوانوما، مننژیوما و اپندیموما میشود.
SWS (جهش موزاییک سوماتیک GNAQ): ناهنجاری در انتقال سیگنال گذرنده وابسته به پروتئین G. زمان و محل جهش در جنین الگوی بیان را در ناحیه عصب سهقلو، داخل جمجمه و چشم متفاوت میکند1). GNAQ همچنین پایه مولکولی مشترک SWS، KTS و PPV است3).
AT (کیناز ATM): عامل سرکوبگر تومور درگیر در ترمیم شکست دو رشتهای DNA و حفظ پایداری ژنوم. جهش → اختلال در ترمیم DNA → استعداد سرطان، افزایش قابل توجه حساسیت به پرتو و نقص ایمنی1).
Chevalier و همکاران (2021) ارتباط بین فاکوماتوزها و تومورهای غدد درونریز را بررسی کردند و گزارش دادند که مسیرهای سیگنالینگ NF1، TSC و VHL (مسیر RAS-PI3K-Akt-mTOR و مسیر HIF) به طور مشترک در ایجاد تومورهای متعدد غدد درونریز نقش دارند2).
Qچرا فاکوماتوزها باعث ایجاد ضایعات در اندامهای متعدد میشوند؟
A
زمینه پاتوفیزیولوژیک مشترک فاکوماتوزها، ناهنجاری در تشکیل، مهاجرت و تمایز سلولهای تاج عصبی است. سلولهای تاج عصبی از اکتودرم منشأ گرفته و سلولهای متنوعی مانند سلولهای شوان و ملانوسیتها را تولید میکنند، بنابراین ضایعات در اندامهای متعدد عصبی، پوستی و چشمی ایجاد میشود. علاوه بر این، جهش در مسیرهای سیگنالینگ مشترک مانند مسیر RAS-mTOR و مسیر VHL-HIF باعث ایجاد تومور در اندامهای مختلف میشود.
7. تحقیقات جدید و چشمانداز آینده (گزارشهای مرحله تحقیقاتی)
این یک داروی هدفمند مولکولی است که توسط FDA برای نوروفیبروم پلکسیفرم غیرقابل جراحی تأیید شده است. این دارو فعالیت بیش از حد مسیر MAPK در NF1 را هدف قرار میدهد 1).
یک مطالعه فاز 2 برای کارسینوم سلول کلیوی سلول شفاف مرتبط با VHL و تومورهای نورواندوکرین پانکراس در حال انجام است.
در مطالعه MK6482 توسط Chevalier و همکاران (2021)، پاسخ عینی در 64% تومورهای نورواندوکرین پانکراس مشاهده شد و بقای بدون پیشرفت در 12 ماه 98.3% گزارش شد2).
در مطالعه EXIST بر روی آنژیومیولیپوم کلیوی و SEGA مرتبط با TSC، پاسخ مورفولوژیک تأیید شده است و انتظار میرود که در تومورهای اندوکرین نیز کاربرد داشته باشد2).
مشخص شده است که جهش ژن GNAQ یک پایه مولکولی مشترک در سندرمهای SWS، Klippel-Trenaunay (KTS) و فاکوماتوز پیگمانوواسکولار (PPV) است3). در این موارد همپوشانی، خطر ملانوم کوروئید وجود دارد، بنابراین معاینه فوندوسکوپی با مردمک گشاد شده 1-2 بار در سال توصیه میشود3).
Gama SM, Tamanini JVG, Moraes MPM, Silva TYT, Lima FT, Pedroso JL, et al. A diagnostic approach to neurocutaneous syndromes. Arquivos de neuro-psiquiatria. 2025;83(7):1-14. doi:10.1055/s-0045-1809664. PMID:40562378; PMCID:PMC12196566.
Chevalier B, Dupuis H, Jannin A, Lemaitre M, Do Cao C, Cardot-Bauters C, et al. Phakomatoses and Endocrine Gland Tumors: Noteworthy and (Not so) Rare Associations. Frontiers in endocrinology. 2021;12:678869. doi:10.3389/fendo.2021.678869. PMID:34025587; PMCID:PMC8134657.
Senthilkumar VA, Kohli P, Mishra C, et al. Ocular features in a patient presenting with a rare combination of multiple phakomatoses. BMJ Case Rep. 2022;15:e252746. doi:10.1136/bcr-2022-252746.
متن مقاله را کپی کنید و در دستیار هوش مصنوعی دلخواه خود بچسبانید.
مقاله در کلیپبورد کپی شد
یکی از دستیارهای هوش مصنوعی زیر را باز کنید و متن کپیشده را در کادر گفتگو بچسبانید.