Bệnh u thần kinh da (phakomatoses) là thuật ngữ chung cho một nhóm bệnh bẩm sinh đặc trưng bởi các tổn thương hamartoma ở da, hệ thần kinh trung ương và mắt. Còn được gọi là hội chứng da thần kinh (neurocutaneous syndromes).
Tên gọi do bác sĩ nhãn khoa người Hà Lan Van der Hoeve đặt, bắt nguồn từ tiếng Hy Lạp «phakos» (thấu kính/đốm). Ban đầu bao gồm ba bệnh: u xơ thần kinh, xơ cứng củ, và bệnh von Hippel-Lindau, sau đó thêm hội chứng Sturge-Weber và mất điều hòa giãn mao mạch. Hiện nay đã mô tả hơn 60 hội chứng.
Cơ sở bệnh lý chung là sự bất thường trong quá trình hình thành, di chuyển và biệt hóa của tế bào mào thần kinh. Tế bào mào thần kinh có nguồn gốc từ ngoại bì và tạo ra nhiều loại tế bào như tế bào Schwann và tế bào hắc tố, dẫn đến tổn thương ở nhiều cơ quan bao gồm thần kinh, da và mắt. Các đường dẫn tín hiệu liên quan bao gồm RAS, MAPK/MEK, mTOR, PI3K/AKT, GNAQ và VHL-HIF.
Sáu bệnh chính là NF1, NF2, xơ cứng củ, hội chứng Sturge-Weber, bệnh von Hippel-Lindau và mất điều hòa giãn mao mạch. Tất cả đều do đột biến gen gây ra và tạo tổn thương ở thần kinh, da và mắt. Hiện nay, hơn 60 hội chứng được xếp vào phạm trù bệnh u thần kinh da.
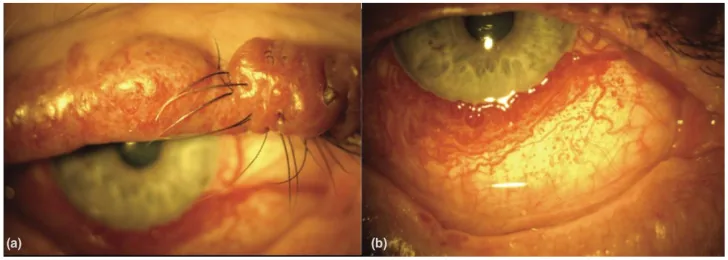
An Update on Multimodal Ophthalmological Imaging of Diffuse Choroidal Hemangioma in Sturge–Weber Syndrome. Vision (Basel). 2023 Oct 6; 7(4):64. Figure 1. PMCID: PMC10594527. License: CC BY.
(a) Vết bớt màu rượu vang ở mí mắt trên có nốt sần ở bệnh nhân hội chứng Sturge–Weber. (b) Mạch máu kết mạc lan tỏa ở bệnh nhân hội chứng Sturge–Weber. Từ [15].
Các loại biến chứng mắt khác nhau ở mỗi bệnh, và triệu chứng chủ quan cũng đa dạng.
NF1: Khi khối u đường thị giác (u thần kinh đệm thị giác) tiến triển, xảy ra giảm thị lực, mất thị giác màu và khiếm khuyết thị trường. U sợi thần kinh đám rối có thể gây lồi mắt.
Xơ cứng củ: U mô thừa tế bào hình sao võng mạc thường không triệu chứng. Gây rối loạn thị lực khi ảnh hưởng đến hoàng điểm hoặc đĩa thị giác.
SWS: Khi kèm glôcôm bẩm sinh, xảy ra đục giác mạc, chảy nước mắt và sợ ánh sáng. Trong glôcôm khởi phát muộn, xảy ra khiếm khuyết thị trường tiến triển không đau.
VHL: Không có triệu chứng ở giai đoạn đầu. Khi u nguyên bào mạch máu mao mạch võng mạc tiến triển, xuất hiện các thay đổi tiết dịch, phù hoàng điểm và vòng trắng, dẫn đến giảm thị lực.
Nốt Lisch: Dấu hiệu mắt thường gặp nhất trong NF1. Các nốt nhỏ (1-2 mm) màu nâu nhạt, ranh giới rõ, hình vòm, nhiều nốt trên mống mắt. Tỷ lệ mắc theo tuổi: <5% ở <3 tuổi, 42% ở 3-4 tuổi, 55% ở 5-6 tuổi, 100% ở >21 tuổi. Được đưa vào tiêu chuẩn chẩn đoán NF1 (≥2 nốt). Vì mống mắt của người Nhật có màu nâu, nên khám bằng đèn khe là quan trọng.
U đường thị giác (u thần kinh đệm thị giác): Xảy ra ở khoảng 15-25% bệnh nhân NF1. Thường là u sao bào lông độ thấp, thường không triệu chứng. Khi tiến triển, gây teo thần kinh thị giác, suy giảm thị lực và khiếm khuyết thị trường. Có thể xâm lấn giao thoa thị giác.
U xơ thần kinh đám rối: Xảy ra ở dưới 10% bệnh nhân NF1. Đặc trưng bởi biến dạng mí mắt hình chữ S, khi sờ có cảm giác như túi giun. Có thể gây lồi mắt, lác, nhược thị và glôcôm bẩm sinh.
Loạn sản cánh lớn xương bướm: Khuyết tật bẩm sinh của thành hốc mắt có thể gây lồi mắt theo nhịp mạch.
Glôcôm: Xảy ra ở 1-2% bệnh nhân NF1. Có hai loại: bẩm sinh (một mắt) và khởi phát muộn.
Khác: Nổi rõ dây thần kinh có myelin ở giác mạc, dày màng mạch và sợi thần kinh có myelin ở võng mạc.
U màng não baodây thần kinh thị giác: Dấu hiệu đặc trưng của NF2. Là loại u tương phản với u thần kinh đệm thị giác trong NF1.
Đục thủy tinh thể: Có thể thấy đục thủy tinh thể ở người trẻ.
U mô thừa võng mạc/biểu mô sắc tố võng mạc, Màng trên võng mạc: Có thể kết hợp.
Viêm giác mạc do lộ: Do rối loạn chức năng dây thần kinh sọ V và VII do u dây thần kinh thính giác hai bên, gây tê mặt, song thị và mắt không nhắm kín.
U mô thừa tế bào hình sao võng mạc: Kết hợp trong khoảng 50% trường hợp. Được phân loại thành 3 loại: (1) phẳng, trong mờ, không vôi hóa, (2) nhô cao, đa thùy, vôi hóa (dạng quả dâu tằm), (3) loại chuyển tiếp. Thường có nhiều u ở cực sau.
Bất thường mạch máu võng mạc: Giãn dạng phình mạch và dị dạng động tĩnh mạch, có thể gây xuất huyết dịch kính, bệnh võng mạc tăng sinh và bong võng mạc.
Khác: U xơ mạch mi mắt, đốm mất sắc tố mống mắt, u mạch màng bồ đào không điển hình.
Tam chứng chính là (1) u máu mặt ở vùng dây thần kinh sinh ba, (2) u máu nội sọ cùng bên, (3) glôcôm hoặc u máu hắc mạc cùng bên.
Glôcôm: Biểu hiện mắt quan trọng nhất trong SWS. Xảy ra ở 30-70% trường hợp. Glôcôm bẩm sinh (từ sơ sinh đến 4 tuổi) chiếm khoảng 60%, gây mắt bò, đục giác mạc và giác mạc to. Nguyên nhân được cho là do bất thường phát triển góc tiền phòng, tăng áp lực tĩnh mạch thượng củng mạc và u máu hắc mạc. Thường xảy ra khi có u máu mi mắt.
U máu màng bồ đào: Xảy ra ở khoảng 20-70% trường hợp. Khó xác định khi khám đáy mắt thông thường vì lan tỏa và ranh giới không rõ. Đáy mắt có dạng như sốt cà chua. Thường không có xu hướng phát triển, nhưng có thể gây thay đổi tiết dịch hoặc bong võng mạc tiết dịch.
Khác: Giãn và ngoằn ngoèo mạch máu kết mạc, thượng củng mạc và mống mắt.
U nguyên bào mạch máu mao mạch võng mạc (u mạch): Xảy ra ở 43-85% bệnh nhân VHL (khoảng 60% trong báo cáo trong nước). Khoảng một phần ba là hai bên và nhiều ổ. Thường gặp ở vùng thái dương ngoại vi võng mạc dưới dạng nốt đỏ cam kèm mạch máu đến và đi giãn ngoằn ngoèo. Tuổi khởi phát trung bình là 25, thường xuất hiện trước 30 tuổi.
Khi tiến triển, gây thay đổi tiết dịch, xuất huyết võng mạc → phù hoàng điểm, vòng trắng → giảm thị lực.
Giãn mao mạch kết mạc nhãn cầu: Dấu hiệu mắt phổ biến nhất. Thường xuất hiện ở độ tuổi 5–8, gặp ở 80–90% bệnh nhân1).
Rối loạn vận động mắt: Rung giật nhãn cầu, mất điều hòa vận nhãn, bất thường vận động nhảy mắt, bất thường hội tụ và điều tiết, và lác.
Thị lực thường được duy trì.
QCác nốt Lisch thay đổi như thế nào theo tuổi?
A
Các nốt Lisch liên quan đến NF1 tăng tần suất xuất hiện theo tuổi. Ở trẻ dưới 3 tuổi, chỉ có 5% có nốt, nhưng ở người trên 21 tuổi, gần như 100% có nốt. Ở người trẻ, có thể khó phát hiện ngay cả bằng đèn khe, do đó cần giải thích dựa trên độ tuổi.
NF1: Khoảng 50% là đột biến de novo. Tỷ lệ thâm nhập gần như hoàn toàn nhưng kiểu hình đa dạng. Neurofibromin hoạt động như RAS-GAP, chuyển GTP-RAS thành GDP-RAS2).
NF2: Mất chức năng protein merlin. Là chất ức chế khối u biểu hiện chủ yếu ở tế bào Schwann và tế bào màng não; thiếu hụt dẫn đến u schwann, u màng não và u tế bào nội tủy.
TSC: Khoảng 2/3 trường hợp là lẻ tẻ. Đột biến TSC2 chiếm 75-80% trường hợp lẻ tẻ và có kiểu hình nặng hơn đột biến TSC11). Hamartin/tuberin ức chế trực tiếp con đường mTOR2).
SWS: Lẻ tẻ do đột biến thể khảm soma ở gen GNAQ1). Vết bớt màu rượu vang (PWB) bao phủ toàn bộ vùng nhánh đầu tiên của dây thần kinh sinh ba (V1) có nguy cơ cao gây biến chứng nhãn khoa và thần kinh.
VHL: Tỷ lệ thâm nhập gần như 100%, khoảng 20% là đột biến de novo. pVHL tham gia vào quá trình ubiquitin hóa và phân hủy HIF-α qua proteasome2). Loại 1 (không u tế bào ưa crom) khoảng 80%, Loại 2 (có u tế bào ưa crom) khoảng 20%2).
AT: Di truyền lặn trên nhiễm sắc thể thường, kinase ATM tham gia sửa chữa đứt gãy chuỗi kép DNA và duy trì ổn định bộ gen 1). Nguy cơ khối u ác tính (hệ bạch huyết) và nhạy cảm với bức xạ rất cao.
Tiêu chuẩn chẩn đoán NF1 (tiêu chuẩn NIH và Hội Da liễu Nhật Bản 2008) quy định rằng chẩn đoán xác định được đưa ra khi đáp ứng hai hoặc nhiều hơn trong số bảy tiêu chí sau.
6 hoặc nhiều hơn các đốm café-au-lait (trước tuổi dậy thì: đường kính ≥5 mm, sau tuổi dậy thì: ≥15 mm)
Tiêu chuẩn lâm sàng: U schwannoma tiền đình hai bên là dấu hiệu đặc trưng. Chẩn đoán có thể khó hơn NF1 do triệu chứng da ít. Xét nghiệm di truyền phát hiện hơn 90% đột biến. CT và MRI xác nhận u thần kinh thính giác hai bên.
Chẩn đoán xác định khi có 2 dấu hiệu chính, hoặc 1 dấu hiệu chính + 2 dấu hiệu phụ trở lên. Việc xác định đột biến gây bệnh TSC1/TSC2 được công nhận là tiêu chuẩn chẩn đoán độc lập 1).
Để đánh giá u tế bào hình sao võng mạc, sử dụng khám đáy mắt, chụp mạch huỳnh quang và OCT. Chẩn đoán phân biệt quan trọng nhất là u nguyên bào võng mạc. Có thể phân biệt dựa trên đặc điểm không vôi hóa ở trẻ em, ít mạch nuôi và có triệu chứng toàn thân, nhưng khó phân biệt với u dâu tằm.
Theo thang chẩn đoán Roach, tiêu chuẩn chẩn đoán là có ít nhất hai trong ba dấu hiệu: bớt đỏ mặt, tăng nhãn áp và u mạch màng não mềm 1). ED-OCT và MRI hữu ích trong đánh giá u mạch màng mạch. CT đầu cho thấy vôi hóa trong vỏ não.
Để theo dõi bệnh tăng nhãn áp, cần thực hiện đo nhãn áp, đánh giá thần kinh thị giác và kiểm tra thị trường một cách định kỳ. Ở trẻ sơ sinh và trẻ nhỏ, ngoài khám bằng đèn khe, có thể cần khám đáy mắt và đo nhãn áp dưới gây mê.
Xét nghiệm di truyền có thể phát hiện đột biến VHL ở gần 100% trường hợp và hiện là phương pháp chính để chẩn đoán xác định. Khám đáy mắt giãn đồng tử được khuyến cáo thực hiện hàng năm từ 1 tuổi. Khối u hình cầu màu đỏ cam ở ngoại vi kèm mạch máu giãn và ngoằn ngoèo là dấu hiệu đặc trưng. Chụp mạch huỳnh quang hữu ích để phát hiện u nguyên bào mạch máu ngoại vi nhỏ, và OCT được sử dụng để đánh giá các tổn thương nhỏ.
Trong chẩn đoán lâm sàng, bộ ba triệu chứng mất điều hòa, giãn mao mạch kết mạc nhãn cầu và bất thường vận động mắt là quan trọng. Các xét nghiệm bao gồm tăng AFP huyết thanh, tăng CA125 và thiếu hụt protein ATM (Western blot). Giãn mao mạch kết mạc nhãn cầu là dấu hiệu bệnh lý đặc hiệu (pathognomonic) của bệnh này. MRI cho thấy teo tiểu não lan tỏa ở hố sau (đặc biệt là thùy nhộng và bán cầu) 1).
Nốt Lisch: U mô thừa không triệu chứng, không cần điều trị.
U thần kinh đệm thị giác: Trong trường hợp tiến triển, cân nhắc phẫu thuật cắt bỏ, nhưng chức năng thị giác bị mất và biến chứng sau phẫu thuật nhiều. Trường hợp xâm lấn giao thoa thị giác, hóa trị liệu được chỉ định.
U sợi thần kinh dạng đám rối: Phẫu thuật cắt bỏ hoàn toàn khó và dễ tái phát. Trường hợp tiến triển, có thể cần phẫu thuật nội soi hốc mắt. Trường hợp không thể cắt bỏ, selumetinib (thuốc ức chế MEK) được sử dụng như liệu pháp mới 1).
Glôcôm: Quản lý theo điều trị glôcôm thông thường.
Đối với u schwann tiền đình, phẫu thuật cắt bỏ là cơ bản; với khối u <3 cm, có thể bảo tồn thính lực trong 65% trường hợp 1). Viêm giác mạc do lộ được xử trí bằng nước mắt nhân tạo, kính bảo vệ giác mạc và khâu mi nếu cần.
Động kinh: Đối với cơn co giật cục bộ ở trẻ nhũ nhi và co thắt trẻ nhũ nhi, vigabatrin là lựa chọn đầu tay1). U sao hình tế bào khổng lồ dưới màng nội tủy (SEGA) là chỉ định cắt bỏ bằng phẫu thuật thần kinh1).
Điều trị glôcôm khác nhau tùy theo thời điểm khởi phát.
Glôcôm bẩm sinh (khởi phát sớm): Phẫu thuật là bắt buộc. Phẫu thuật cắt bè củng mạc và phẫu thuật mở góc tiền phòng được lựa chọn. Cần chú ý đầy đủ đến bong hắc mạc và xuất huyết, nguy cơ biến chứng cao hơn bình thường do tăng áp lực tĩnh mạch thượng củng mạc. Có xu hướng kháng với điều trị nội khoa.
Thể khởi phát muộn: Đầu tiên điều trị bằng thuốc, nếu không hiệu quả thì xem xét phẫu thuật.
Điều trị u máu màng mạch được lựa chọn dựa trên sự hiện diện của các thay đổi tiết dịch từ các lựa chọn sau:
Nếu có thay đổi tiết dịch → quang đông
Quang đông không hiệu quả hoặc bong võng mạc tiết dịch → xạ trị (tổng liều khoảng 20 Gray). Có thể kỳ vọng làm phẳng bong võng mạc dạng bọng và thu nhỏ u máu.
Quang đông (lựa chọn đầu tiên): Sử dụng laser argon hoặc laser nhuộm, đông đặc võng mạc xung quanh u mạch và các mạch nuôi, sau đó đông trực tiếp khối u. Đối với khối u có đường kính ≥2 đĩa thị, đông động mạch đến và võng mạc xung quanh trước, sau đó đông trực tiếp khối u.
Đông lạnh: Thực hiện qua củng mạc trong trường hợp u mạch lớn hoặc ở vùng ngoại vi võng mạc khó quang đông.
Phẫu thuật cắt dịch kính: Được xem xét khi có thay đổi tiết dịch nặng, bong võng mạc hoặc thay đổi tăng sinh.
Giai đoạn cuối: Cần quản lý bệnh tăng nhãn áptân mạch.
Nếu có tiền sử gia đình, hãy khám đáy mắt cho tất cả thành viên; tiên lượng tốt nếu điều trị sớm.
Không có điều trị đặc hiệu cho giãn mao mạch kết mạc. Đối với suy giảm miễn dịch, dùng kháng sinh dự phòng và immunoglobulin tĩnh mạch (IVIg)1). Mất điều hòa được điều trị triệu chứng.
QKhi nào nên tầm soát u máu võng mạc trong VHL?
A
Trong bệnh VHL, khuyến cáo khám đáy mắt giãn đồng tử mỗi năm một lần từ 1 tuổi. Tất cả các thành viên trong gia đình bệnh nhân có đột biến gen VHL đã được xác nhận cũng cần được khám đáy mắt, vì điều trị sớm có thể mang lại tiên lượng chức năng thị giác tốt.
Cơ chế bệnh sinh ở mức phân tử của từng bệnh được trình bày dưới đây.
NF1 và TSC (Con đường mTOR)
NF1 (Neurofibromin): Hoạt động như RAS-GAP. Thúc đẩy chuyển đổi RAS gắn GTP (dạng hoạt động) thành RAS gắn GDP (dạng không hoạt động). Đột biến dẫn đến kích hoạt RAS liên tục, mất kiểm soát con đường MAP kinase và PI3K-Akt-mTOR, gây tăng sinh tế bào không kiểm soát 2).
TSC (Hamartin/Tuberin): Phức hợp ức chế khối u trực tiếp ức chế phức hợp mTOR 1 và 2. Đột biến dẫn đến hoạt động quá mức của mTOR, gây bất thường trong chuyển hóa năng lượng, tổng hợp protein/lipid và sự sống của tế bào 2). Đột biến TSC2 gây kiểu hình nặng hơn đột biến TSC1 1).
VHL (con đường HIF)
pVHL: Thành phần của phức hợp E3 ubiquitin ligase (elongin B/C, Cullin 2, RBX1). Trong điều kiện oxy bình thường, prolyl hydroxylase hydroxyl hóa HIF-α → pVHL nhận diện → ubiquitin hóa → phân hủy proteasome.
Đột biến VHL: Tình trạng giả thiếu oxy diễn ra liên tục dẫn đến tích tụ HIF-α, hình thành dị vòng với HIF-1β, kích hoạt phiên mã các gen liên quan đến VEGF, sản xuất hồng cầu, trao đổi chất và tăng sinh tế bào, gây ra sự hình thành khối u mạch máu 2).
NF2 (Merlin) : protein ức chế khối u biểu hiện ở tế bào Schwann và tế bào màng não. Đột biến gây u schwann, u màng não và u tế bào nội tủy.
SWS (đột biến thể khảm soma GNAQ) : bất thường trong truyền tín hiệu xuyên màng liên quan đến protein G. Mô hình biểu hiện ở vùng dây thần kinh sinh ba, nội sọ và mắt khác nhau tùy thuộc vào thời điểm và vị trí đột biến trong phôi 1). GNAQ cũng là nền tảng phân tử chung cho SWS, KTS và PPV3).
AT (ATM kinase): Yếu tố ức chế khối u tham gia sửa chữa đứt gãy chuỗi kép DNA và duy trì ổn định bộ gen. Đột biến → suy giảm sửa chữa DNA → dễ mắc ung thư, tăng nhạy cảm với bức xạ đáng kể và suy giảm miễn dịch1).
Chevalier và cộng sự (2021) đã nghiên cứu mối liên quan giữa bệnh u thần kinh da và khối u nội tiết, và báo cáo rằng các đường dẫn tín hiệu (RAS-PI3K-Akt-mTOR, HIF) trong NF1, TSC và VHL có liên quan chung đến sự phát triển của khối u nội tiết đa ổ2).
QTại sao bệnh u thần kinh da lại gây tổn thương ở nhiều cơ quan?
A
Cơ sở bệnh lý chung của bệnh u thần kinh da là sự bất thường trong quá trình hình thành, di chuyển và biệt hóa của tế bào mào thần kinh. Tế bào mào thần kinh có nguồn gốc từ ngoại bì và tạo ra nhiều loại tế bào như tế bào Schwann và tế bào hắc tố, do đó tổn thương lan rộng đến nhiều cơ quan bao gồm thần kinh, da và mắt. Ngoài ra, các đột biến trong đường dẫn tín hiệu chung như RAS-mTOR và VHL-HIF thúc đẩy sự phát triển khối u vượt qua các cơ quan.
7. Nghiên cứu mới nhất và triển vọng tương lai (báo cáo giai đoạn nghiên cứu)
Thuốc nhắm mục tiêu phân tử được FDA phê duyệt cho u xơ thần kinh dạng đám rối không thể phẫu thuật cắt bỏ. Nhắm vào sự hoạt động quá mức của con đường MAPK trong NF11).
Một thử nghiệm lâm sàng đang được tiến hành tại liên minh INTUITT-NF2 cho u schwannoma liên quan đến NF2 và khối u tiến triển, với kết quả khả quan đã được báo cáo 1).
Một thử nghiệm giai đoạn 2 cho ung thư biểu mô tế bào thận trong suốt liên quan đến VHL và khối u thần kinh nội tiết tụy đang được tiến hành.
Trong thử nghiệm MK6482 của Chevalier và cộng sự (2021), 64% khối u thần kinh nội tiết tụy có đáp ứng khách quan, tỷ lệ sống không tiến triển sau 12 tháng là 98,3% 2).
Thuốc ức chế mTOR everolimus (khối u liên quan đến TSC)
Thử nghiệm EXIST đối với u mỡ cơ mạch thận liên quan đến TSC và SEGA đã xác nhận đáp ứng hình thái, và ứng dụng cho các khối u nội tiết cũng được kỳ vọng 2).
Đột biến gen GNAQ được phát hiện là cơ sở phân tử chung cho SWS, hội chứng Klippel-Trenaunay (KTS) và bớt mạch máu sắc tố (PPV)3). Trong các trường hợp chồng lấn này, có nguy cơ u hắc tố màng mạch, do đó khuyến cáo khám đáy mắt với giãn đồng tử 1-2 lần mỗi năm3).
Gama SM, Tamanini JVG, Moraes MPM, Silva TYT, Lima FT, Pedroso JL, et al. A diagnostic approach to neurocutaneous syndromes. Arquivos de neuro-psiquiatria. 2025;83(7):1-14. doi:10.1055/s-0045-1809664. PMID:40562378; PMCID:PMC12196566.
Chevalier B, Dupuis H, Jannin A, Lemaitre M, Do Cao C, Cardot-Bauters C, et al. Phakomatoses and Endocrine Gland Tumors: Noteworthy and (Not so) Rare Associations. Frontiers in endocrinology. 2021;12:678869. doi:10.3389/fendo.2021.678869. PMID:34025587; PMCID:PMC8134657.
Senthilkumar VA, Kohli P, Mishra C, et al. Ocular features in a patient presenting with a rare combination of multiple phakomatoses. BMJ Case Rep. 2022;15:e252746. doi:10.1136/bcr-2022-252746.
Sao chép toàn bộ bài viết và dán vào trợ lý AI bạn muốn dùng.
Đã sao chép bài viết vào clipboard
Mở một trợ lý AI bên dưới và dán nội dung đã sao chép vào ô chat.