Fakomatosis (phakomatoses) adalah istilah umum untuk sekelompok penyakit bawaan yang ditandai dengan lesi hamartoma pada kulit, sistem saraf pusat, dan mata. Juga disebut sindrom neurokutaneus (neurocutaneous syndromes).
Penamaan berasal dari dokter mata Belanda Van der Hoeve, dari bahasa Yunani «phakos» (lensa/bintik). Awalnya mencakup tiga penyakit: neurofibromatosis, sklerosis tuberosa, dan penyakit von Hippel-Lindau, kemudian ditambahkan sindrom Sturge-Weber dan ataksia telangiektasis. Saat ini lebih dari 60 sindrom telah dideskripsikan.
Dasar patofisiologi yang umum adalah kelainan pada pembentukan, migrasi, dan diferensiasi sel krista neural. Sel krista neural berasal dari ektoderm dan menghasilkan berbagai sel seperti sel Schwann dan melanosit, sehingga menyebabkan lesi pada banyak organ termasuk saraf, kulit, dan mata. Jalur sinyal yang terlibat meliputi RAS, MAPK/MEK, mTOR, PI3K/AKT, GNAQ, dan VHL-HIF.
Frekuensi kejadian 6 penyakit utama adalah sebagai berikut:
QPenyakit apa saja yang termasuk dalam phakomatosis?
A
Enam penyakit utama adalah NF1, NF2, sklerosis tuberosa, sindrom Sturge-Weber, penyakit von Hippel-Lindau, dan ataksia telangiektasia. Semuanya disebabkan oleh mutasi genetik dan menyebabkan lesi pada saraf, kulit, dan mata. Saat ini, lebih dari 60 sindrom termasuk dalam kategori fakomatosis.
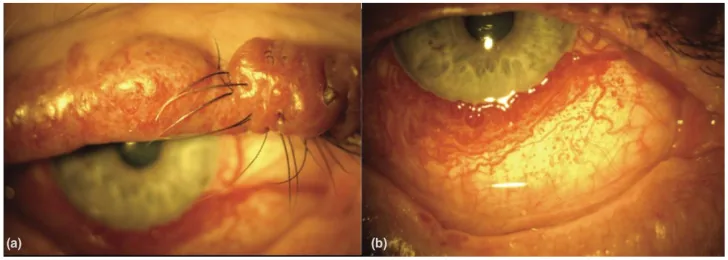
An Update on Multimodal Ophthalmological Imaging of Diffuse Choroidal Hemangioma in Sturge–Weber Syndrome. Vision (Basel). 2023 Oct 6; 7(4):64. Figure 1. PMCID: PMC10594527. License: CC BY.
(a) Noda port-wine stain pada kelopak atas pada pasien sindrom Sturge–Weber. (b) Vaskularitas konjungtiva difus pada pasien sindrom Sturge–Weber. Dari [15].
Jenis komplikasi okular berbeda pada setiap penyakit, dan gejala subjektif juga bervariasi.
NF1: Ketika tumor jalur optik (glioma optik) berkembang, terjadi penurunan ketajaman penglihatan, kehilangan penglihatan warna, dan defek lapang pandang. Neurofibroma pleksiformis dapat menyebabkan proptosis.
Sklerosis tuberosa: Hamartoma sel bintang retina biasanya asimtomatik. Gangguan penglihatan terjadi jika mengenai makula atau diskus optikus.
SWS: Jika disertai glaukoma kongenital, terjadi kekeruhan kornea, lakrimasi, dan fotofobia. Pada glaukoma onset lambat, terjadi defek lapang pandang progresif tanpa nyeri.
VHL: Awalnya tanpa gejala. Ketika hemangioblastoma retina berkembang, muncul perubahan eksudatif, edema makula, dan lingkaran putih, menyebabkan penurunan penglihatan.
Nodul Lisch: Temuan okular paling sering pada NF1. Nodul kecil (1-2 mm) berwarna coklat muda, batas tegas, berbentuk kubah, multipel pada iris. Prevalensi berdasarkan usia: <5% pada usia <3 tahun, 42% pada usia 3-4 tahun, 55% pada usia 5-6 tahun, 100% pada usia >21 tahun. Termasuk dalam kriteria diagnosis NF1 (≥2 nodul). Karena iris orang Jepang berwarna coklat, pemeriksaan dengan slit-lamp penting.
Tumor jalur optik (glioma optik): Terjadi pada sekitar 15-25% pasien NF1. Sering berupa astrositoma piloidik derajat rendah, sering asimtomatik. Jika progresif, menyebabkan atrofi saraf optik, gangguan penglihatan, dan defek lapang pandang. Dapat menginvasi kiasma optikum.
Neurofibroma pleksiformis: Terjadi pada kurang dari 10% pasien NF1. Ditandai dengan deformitas kelopak mata berbentuk S, dan pada palpasi teraba seperti kantong berisi cacing. Dapat menyebabkan proptosis, strabismus, ambliopia, dan glaukoma kongenital.
Displasia sayap besar tulang sfenoid: Defek kongenital dinding orbita yang dapat menyebabkan proptosis pulsatif.
Glaukoma: Terjadi pada 1-2% pasien NF1. Ada dua tipe: kongenital (unilateral) dan onset lambat.
Hamartoma retina/RPE, Membran epiretina: Dapat menyertai.
Keratitis eksposur: Disebabkan oleh disfungsi saraf kranial V dan VII akibat neuroma akustik bilateral, yang menyebabkan mati rasa wajah, diplopia, dan lagoftalmus.
Hamartoma astrositik retina: Terjadi pada sekitar 50% kasus. Diklasifikasikan menjadi 3 tipe: (1) datar, tembus cahaya, non-kalsifikasi, (2) meninggi, multinodular, kalsifikasi (penampilan seperti murbei), (3) tipe transisional. Biasanya multipel di kutub posterior.
Lesi retina depigmentasi: Tampilan seperti “berlubang” (punched-out).
Trias utama adalah (1) hemangioma wajah di area saraf trigeminus, (2) hemangioma intrakranial ipsilateral, (3) glaukoma atau hemangiomakoroid ipsilateral.
Glaukoma: Temuan okular terpenting pada SWS. Terjadi pada 30-70% kasus. Glaukoma kongenital (lahir hingga 4 tahun) mencakup sekitar 60%, menyebabkan buftalmos, kekeruhan kornea, dan megalokornea. Etiologi diduga melibatkan disgenesis sudut, peningkatan tekanan vena episklera, dan hemangiomakoroid. Sering terjadi dengan adanya hemangioma palpebra.
Hemangiomakoroid: Terjadi pada sekitar 20-70% kasus. Sulit diidentifikasi pada pemeriksaan fundus biasa karena difus dan batas tidak jelas. Fundus tampak seperti saus tomat. Biasanya tidak cenderung membesar, tetapi dapat menyebabkan perubahan eksudatif atau ablasi retina eksudatif.
Lainnya: Pelebaran dan tortuositas pembuluh darah konjungtiva, episklera, dan iris.
Temuan okular pada VHL (Penyakit Von Hippel-Lindau)
Hemangioblastoma kapiler retina (angioma): Terjadi pada 43-85% pasien VHL (sekitar 60% dalam laporan domestik). Sekitar sepertiga bilateral dan multipel. Sering ditemukan di daerah temporal perifer retina sebagai nodul merah-oranye dengan pembuluh darah aferen dan eferen yang melebar dan berkelok-kelok. Usia rata-rata onset adalah 25 tahun, biasanya muncul sebelum usia 30.
Perkembangan lanjut menyebabkan perubahan eksudatif, perdarahan retina → edema makula, lingkaran putih → penurunan visus.
Pada angiografi fluorescein, terlihat aliran zat warna ke arteri aferen → vena eferen, dengan kebocoran zat warna yang jelas dari massa tumor pada fase awal.
Telangiektasis konjungtiva bulbar: Temuan okular paling umum. Biasanya muncul pada usia 5–8 tahun, ditemukan pada 80–90% pasien1).
Gangguan gerakan mata: Nistagmus, apraksia okulomotor, abnormalitas sakad, abnormalitas konvergensi dan akomodasi, serta strabismus.
Ketajaman penglihatan biasanya tetap terjaga.
QBagaimana nodus Lisch berubah seiring bertambahnya usia?
A
Nodul Lisch yang terkait dengan NF1 meningkat frekuensinya seiring bertambahnya usia. Pada usia di bawah 3 tahun, hanya 5% yang ditemukan, tetapi pada usia di atas 21 tahun, hampir 100% terdeteksi. Pada usia muda, mungkin sulit dideteksi bahkan dengan slit lamp, sehingga interpretasi harus mempertimbangkan usia.
Karakteristik genetik dari setiap penyakit adalah sebagai berikut:
NF1: Sekitar 50% adalah mutasi de novo. Penetrasi hampir sempurna tetapi fenotip bervariasi. Neurofibromin berfungsi sebagai RAS-GAP, mengubah GTP-RAS menjadi GDP-RAS2).
NF2: Kehilangan fungsi protein merlin. Merupakan penekan tumor yang diekspresikan terutama pada sel Schwann dan sel meningeal; defisiensi menyebabkan schwannoma, meningioma, dan ependymoma.
TSC: Sekitar 2/3 kasus bersifat sporadis. Mutasi TSC2 mencakup 75-80% kasus sporadis dan memiliki fenotip yang lebih parah dibandingkan mutasi TSC11). Hamartin/tuberin secara langsung menghambat jalur mTOR2).
SWS: Sporadis disebabkan oleh mutasi mosaik somatik pada gen GNAQ1). Noda port-wine (PWB) yang menutupi seluruh area cabang pertama saraf trigeminal (V1) berisiko tinggi menyebabkan komplikasi oftalmologis dan neurologis.
VHL: Penetrasi hampir 100%, sekitar 20% mutasi de novo. pVHL terlibat dalam ubiquitinasi dan degradasi HIF-α melalui proteasom2). Tipe 1 (tanpa feokromositoma) sekitar 80%, Tipe 2 (dengan feokromositoma) sekitar 20%2).
AT: Pewarisan resesif autosom, ATM kinase terlibat dalam perbaikan patahan untai ganda DNA dan pemeliharaan stabilitas genom 1). Risiko tumor ganas (limfatik) dan sensitivitas radiasi sangat tinggi.
Kriteria diagnosis NF1 (kriteria NIH dan Perhimpunan Dermatologi Jepang 2008) menyatakan bahwa diagnosis pasti ditegakkan jika memenuhi dua atau lebih dari tujuh item berikut.
6 atau lebih bercak café-au-lait (sebelum pubertas: diameter ≥5 mm, setelah pubertas: ≥15 mm)
2 atau lebih neurofibroma atau neurofibroma difus
Bintik-bintik seperti freckle di ketiak atau selangkangan
Kriteria klinis: Schwannoma vestibular bilateral merupakan temuan khas. Diagnosis mungkin lebih sulit daripada NF1 karena gejala kulit yang minim. Tes genetik mendeteksi lebih dari 90% mutasi. CT dan MRI digunakan untuk mengonfirmasi tumor saraf pendengaran bilateral.
Diagnosis pasti ditegakkan dengan 2 temuan utama, atau 1 temuan utama + 2 atau lebih temuan minor. Identifikasi mutasi patogenik TSC1/TSC2 diakui sebagai kriteria diagnostik independen 1).
Untuk mengevaluasi astrositoma retina, digunakan pemeriksaan fundus, angiografi fluorescein, dan OCT. Diagnosis banding yang paling penting adalah retinoblastoma. Dapat dibedakan berdasarkan tidak adanya kalsifikasi pada masa kanak-kanak, sedikitnya pembuluh darah nutrisi, dan adanya gejala sistemik, namun sulit dibedakan dari tumor murbei.
Pada skala diagnostik Roach, kriteria diagnosis adalah memenuhi setidaknya dua dari tiga: nevus flammeus wajah, peningkatan tekanan intraokular, dan angioma leptomeningeal 1). ED-OCT dan MRI berguna untuk mengevaluasi angioma koroid. CT kepala menunjukkan kalsifikasi intrakortikal serebral.
Untuk pemantauan glaukoma, dilakukan pengukuran tekanan intraokular, evaluasi saraf optik, dan tes lapang pandang secara teratur. Pada bayi dan anak kecil, selain pemeriksaan slit-lamp, mungkin diperlukan pemeriksaan fundus dan pengukuran tekanan intraokular di bawah anestesi.
Tes genetik dapat mendeteksi mutasi VHL pada hampir 100% kasus dan saat ini menjadi metode utama diagnosis definitif. Pemeriksaan fundus dengan pupil dilatasi dianjurkan dilakukan setiap tahun mulai usia 1 tahun. Temuan khas adalah tumor bulat merah-oranye di perifer dengan pembuluh darah melebar dan berkelok-kelok. Angiografi fluorescein berguna untuk mendeteksi hemangioblastoma perifer kecil, dan OCT digunakan untuk mengevaluasi lesi kecil.
Dalam diagnosis klinis, tiga serangkai ataksia, telangiektasis konjungtiva bulbar, dan gangguan gerakan mata sangat penting. Temuan laboratorium meliputi peningkatan AFP serum, peningkatan CA125, dan defisiensi protein ATM (Western blot). Telangiektasis konjungtiva bulbar merupakan temuan patognomonik untuk penyakit ini. MRI menunjukkan atrofi serebelar difus di fossa posterior (terutama vermis dan hemisfer) 1).
Nodul Lisch: Hamartoma asimtomatik yang tidak memerlukan terapi.
Glioma optik: Pada kasus progresif, pertimbangkan reseksi bedah, namun fungsi penglihatan hilang dan komplikasi pasca operasi sering terjadi. Pada kasus infiltrasi kiasma, kemoterapi diindikasikan.
Neurofibroma pleksiformis: Eksisi bedah total sulit dan mudah kambuh. Pada kasus progresif, mungkin diperlukan eviserasiorbita. Pada kasus yang tidak dapat direseksi, selumetinib (inhibitor MEK) digunakan sebagai terapi baru 1).
Glaukoma: Dikelola sesuai dengan tata laksana glaukoma biasa.
Untuk schwannoma vestibular, reseksi bedah adalah dasar; pada tumor <3 cm, pendengaran dapat dipertahankan pada 65% kasus 1). Keratitis eksposur ditangani dengan air mata buatan, kacamata pelindung kornea, dan tarsorafi jika perlu.
Pengobatan terkait mata pada sklerosis tuberosa (TSC)
Astrositoma hamartoma retina: Biasanya tidak menunjukkan kecenderungan membesar dan tidak memerlukan pengobatan.
Kelainan pembuluh darah retina (ektasis aneurisma, malformasi arteriovenosa): Fotokoagulasi profilaksis dilakukan karena risiko perdarahan vitreus dan ablasi retina. Jika terjadi perdarahan vitreus atau ablasi retina, pertimbangkan vitrektomi.
Epilepsi: Untuk kejang parsial infantil dan spasme infantil, vigabatrin adalah pilihan pertama1). SEGA (astrositoma subependimal sel raksasa) merupakan indikasi untuk reseksi bedah saraf1).
Pengobatan glaukoma berbeda tergantung pada waktu onset.
Glaukoma kongenital (onset dini): Perawatan bedah wajib dilakukan. Trabekulotomi dan goniotomi menjadi pilihan. Perlu perhatian penuh terhadap ablasi dan perdarahan koroid, dengan risiko komplikasi lebih tinggi dari biasanya akibat peningkatan tekanan vena episklera. Ada kecenderungan resistensi terhadap pengobatan medikamentosa.
Tipe onset lambat: Pertama, terapi obat dilakukan; jika tidak efektif, pertimbangkan operasi.
Pengobatan hemangiomakoroid dipilih berdasarkan ada tidaknya perubahan eksudatif dari berikut:
Jika terjadi perubahan eksudatif → fotokoagulasi
Fotokoagulasi tidak efektif atau ablasi retina eksudatif → radioterapi (dosis total sekitar 20 Gray). Diharapkan dapat memperbaiki ablasi retina bula dan mengecilkan hemangioma.
Fotokoagulasi (pilihan pertama): Menggunakan laser argon atau laser pewarna, koagulasi padat pada retina di sekitar hemangioma dan pembuluh darah nutrisi, kemudian koagulasi langsung pada tumor. Untuk tumor berdiameter ≥2 diskus optikus, koagulasi arteri aferen dan retina sekitarnya terlebih dahulu, lalu koagulasi langsung tumor.
Kriokoagulasi: Dilakukan secara transskleral pada kasus hemangioma besar atau di perifer retina yang sulit difotokoagulasi.
Vitrektomi: Dipertimbangkan jika terdapat perubahan eksudatif berat, ablasi retina, atau perubahan proliferatif.
Tidak ada pengobatan spesifik untuk telangiektasis konjungtiva. Untuk imunodefisiensi, diberikan antibiotik profilaksis dan imunoglobulin intravena (IVIg)1). Ataksia ditangani secara simtomatik.
QKapan sebaiknya pemeriksaan hemangioma retina pada VHL dimulai?
A
Pada VHL, pemeriksaan fundus dengan dilatasi setahun sekali direkomendasikan mulai usia 1 tahun. Semua anggota keluarga pasien dengan mutasi gen VHL yang terkonfirmasi juga penting untuk menjalani pemeriksaan fundus, karena pengobatan dini dapat memberikan prognosis fungsi visual yang baik.
Mekanisme molekuler masing-masing penyakit dijelaskan di bawah ini.
NF1 dan TSC (Jalur mTOR)
NF1 (Neurofibromin): Berfungsi sebagai RAS-GAP. Mempercepat konversi RAS terikat GTP (bentuk aktif) menjadi RAS terikat GDP (bentuk tidak aktif). Mutasi menyebabkan aktivasi RAS terus-menerus, disregulasi jalur MAP kinase dan PI3K-Akt-mTOR, mengakibatkan proliferasi sel yang tidak terkendali 2).
TSC (Hamartin/Tuberin): Kompleks penekan tumor yang secara langsung menghambat kompleks mTOR 1 dan 2. Mutasi menyebabkan hiperaktivitas mTOR, mengakibatkan abnormalitas metabolisme energi, sintesis protein/lipid, dan kelangsungan hidup sel 2). Mutasi TSC2 menghasilkan fenotipe yang lebih parah daripada mutasi TSC1 1).
VHL (jalur HIF)
pVHL: Komponen kompleks E3 ubiquitin ligase (elongin B/C, Cullin 2, RBX1). Dalam kondisi oksigen normal, prolyl hydroxylase menghidroksilasi HIF-α → dikenali oleh pVHL → ubiquitinasi → degradasi proteasom.
Mutasi VHL: Keadaan pseudo-hipoksia yang terus-menerus menyebabkan akumulasi HIF-α, pembentukan heterodimer dengan HIF-1β, aktivasi transkripsi gen terkait VEGF, produksi eritrosit, metabolisme, dan proliferasi sel, yang mengarah pada pembentukan tumor vaskular 2).
NF2 (Merlin) : protein penekan tumor yang diekspresikan pada sel Schwann dan sel leptomeningeal. Mutasi menyebabkan schwannoma, meningioma, dan ependymoma.
SWS (mutasi mosaik somatik GNAQ) : kelainan sinyal transmembran terkait protein G. Pola ekspresi di area saraf trigeminal, intrakranial, dan mata berbeda tergantung waktu dan lokasi mutasi pada embrio 1). GNAQ juga merupakan dasar molekuler bersama untuk SWS, KTS, dan PPV3).
AT (ATM kinase): Faktor penekan tumor yang terlibat dalam perbaikan patahan untai ganda DNA dan pemeliharaan stabilitas genom. Mutasi → gangguan perbaikan DNA → predisposisi kanker, peningkatan sensitivitas radiasi yang signifikan, dan defisiensi imun1).
Chevalier dkk. (2021) meneliti hubungan antara phakomatosis dan tumor endokrin, dan melaporkan bahwa jalur sinyal NF1, TSC, VHL (jalur RAS-PI3K-Akt-mTOR, jalur HIF) secara umum terlibat dalam terjadinya tumor endokrin multipel2).
QMengapa phakomatosis menyebabkan lesi di banyak organ?
A
Dasar patofisiologi umum phakomatosis adalah kelainan pada pembentukan, migrasi, dan diferensiasi sel krista neuralis. Sel krista neuralis berasal dari ektoderm dan menghasilkan berbagai sel seperti sel Schwann dan melanosit, sehingga lesi meluas ke banyak organ termasuk saraf, kulit, dan mata. Selain itu, mutasi pada jalur sinyal umum seperti RAS-mTOR dan VHL-HIF mendorong terjadinya tumor di berbagai organ.
7. Penelitian terbaru dan prospek masa depan (laporan tahap penelitian)
Uji klinis sedang dilakukan di konsorsium INTUITT-NF2 untuk schwannoma terkait NF2 dan tumor progresif, dengan hasil yang menjanjikan telah dilaporkan 1).
Uji coba fase 2 untuk karsinoma sel ginjal jernih terkait VHL dan tumor neuroendokrin pankreas sedang berlangsung.
Dalam uji coba MK6482 oleh Chevalier dkk. (2021), respons objektif diperoleh pada 64% tumor neuroendokrin pankreas, dan tingkat kelangsungan hidup bebas progresi 12 bulan dilaporkan sebesar 98,3% 2).
Uji EXIST untuk angiomiolipoma ginjal terkait TSC dan SEGA telah mengonfirmasi respons morfologis, dan aplikasi pada tumor endokrin juga diharapkan 2).
Mutasi gen GNAQ ditemukan sebagai dasar molekuler yang umum untuk SWS, sindrom Klippel-Trenaunay (KTS), dan nevus vaskular pigmen (PPV)3). Pada kasus tumpang tindih ini, terdapat risiko melanoma koroid, sehingga disarankan pemeriksaan fundus dengan dilatasi pupil 1-2 kali setahun3).
Gama SM, Tamanini JVG, Moraes MPM, Silva TYT, Lima FT, Pedroso JL, et al. A diagnostic approach to neurocutaneous syndromes. Arquivos de neuro-psiquiatria. 2025;83(7):1-14. doi:10.1055/s-0045-1809664. PMID:40562378; PMCID:PMC12196566.
Chevalier B, Dupuis H, Jannin A, Lemaitre M, Do Cao C, Cardot-Bauters C, et al. Phakomatoses and Endocrine Gland Tumors: Noteworthy and (Not so) Rare Associations. Frontiers in endocrinology. 2021;12:678869. doi:10.3389/fendo.2021.678869. PMID:34025587; PMCID:PMC8134657.
Senthilkumar VA, Kohli P, Mishra C, et al. Ocular features in a patient presenting with a rare combination of multiple phakomatoses. BMJ Case Rep. 2022;15:e252746. doi:10.1136/bcr-2022-252746.
Salin teks artikel dan tempelkan ke asisten AI pilihan Anda.
Artikel disalin ke papan klip
Buka asisten AI di bawah, lalu tempelkan teks yang disalin ke kotak chat.