Glioma saraf optik (optic nerve glioma / optic pathway glioma) adalah jenis glioma yang terjadi pada saraf optik. Dalam arti sempit, ini merujuk pada glioma yang timbul di saraf optik sebelum kiasma optikum. Dalam arti luas, ini berarti glioma yang terjadi di seluruh jalur optik termasuk area di belakang kiasma optikum (optic pathway glioma).
Secara histologis, sebagian besar adalah astrositoma pilocytic jinak (pilocytic astrocytoma, WHO Grade I). Namun, beberapa kasus ganas juga dilaporkan. Sekitar 70% terjadi pada masa kanak-kanak, dan merupakan penyakit langka yang mencakup sekitar 0,5–5% dari tumor otak anak.
Bentuk paling sering terjadi. Gejala utama adalah penurunan penglihatan unilateral dan proptosis.
Terbatas pada saraf optik di dalam orbita, observasi merupakan dasar penanganan. Pada kasus dengan NF1, dilaporkan adanya regresi spontan.
Tipe infiltrasi kiasma optikum
Tipe yang menginfiltrasi hingga ke kiasma optikum.
Menyebabkan gangguan penglihatan bilateral dan penanganannya menjadi kompleks.
Sering terjadi pada usia muda, perlu dievaluasi penyebaran ke hipotalamus.
Tipe Jalur Optik-Hipotalamus
Tipe yang menyebar dari belakang kiasma optikum hingga ke hipotalamus.
Dapat disertai kelainan endokrin (gangguan pertumbuhan, pubertas dini, dll.).
Dalam penanganan, diperlukan kerja sama dengan bedah saraf dan endokrinologi.
Klasifikasi berdasarkan latar belakang genetik meliputi tipe terkait NF1 (sekitar 30% dari total) dan tipe sporadik (sekitar 70%).
Pada tipe terkait NF1, dapat terjadi bilateral.
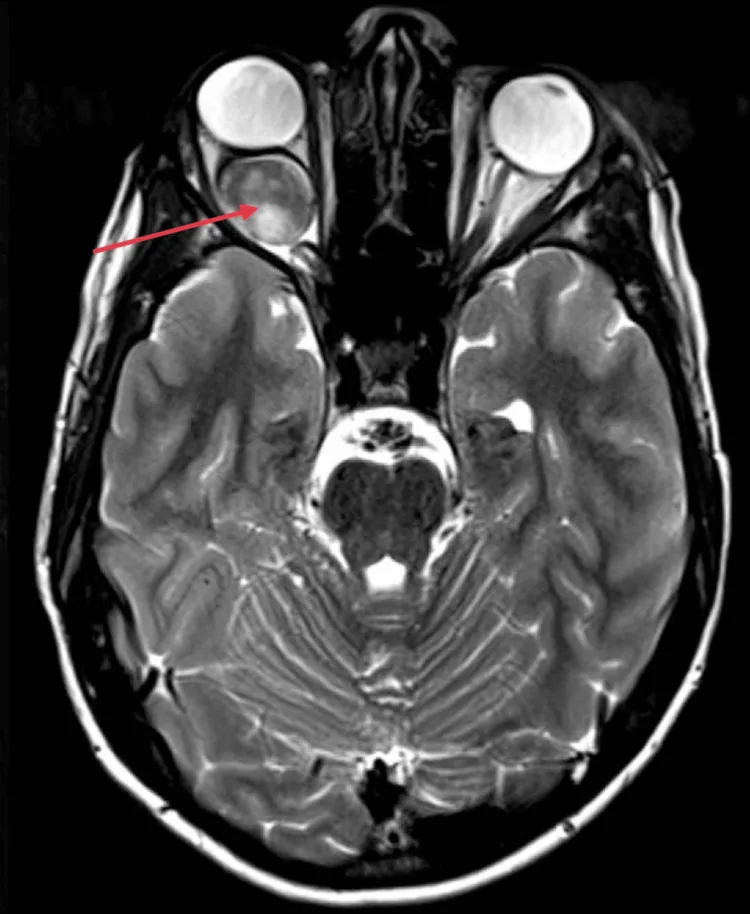
Kotecha MR, et al. Idiopathic Optic Nerve Glioma: A Case Report. Cureus. 2024. Figure 2. PMCID: PMC11744336. License: CC BY.
Pada MRI aksial, saraf optikus intraorbital kanan tampak membesar secara fusiformis, menunjukkan efek kontras yang kuat dan homogen (panah merah), serta proptosis akibat efek massa ke posterior bola mata. Sesuai dengan temuan MRI (pembesaran saraf optikus homogen, downward kinking) yang dibahas pada bagian “2. Gejala utama dan temuan klinis”.
Anak kecil sering tidak mengeluhkan penurunan penglihatan. Oleh karena itu, orang tua atau orang di sekitarnya sering kali menyadari strabismus (terutama esotropia) dan membawanya ke dokter mata untuk pertama kali.
Penurunan penglihatan unilateral tanpa strabismus lebih sulit terdeteksi. Pada kunjungan pertama, mungkin sudah terjadi atrofi saraf optik.
Glioma optik bilateral lebih sering terjadi pada kasus dengan onset usia dini. Pertama kali diketahui karena kelainan arah pandangan atau perilaku yang menunjukkan tidak melihat, dan gangguan penglihatan bisa parah.
Proptosis tidak jelas dan tidak ada nyeri.
QJika anak didiagnosis strabismus, apakah mungkin itu glioma saraf optik?
A
Pada glioma saraf optik, anak kecil sering tidak dapat menyadari atau mengeluhkan penurunan penglihatan, sehingga strabismus (terutama esotropia) sering menjadi gejala awal yang mendorong kunjungan ke dokter mata. Pada anak yang didiagnosis strabismus, terutama strabismus unilateral, penting untuk mempertimbangkan glioma saraf optik dan melakukan pemeriksaan menyeluruh termasuk tes ketajaman penglihatan, fundus, dan pencitraan.
Pada lesi orbitaNF1, glioma optikus adalah yang paling sering
QApakah NF1 (neurofibromatosis tipe 1) meningkatkan risiko glioma optik?
A
Pasien NF1 memiliki risiko glioma optik yang sangat tinggi, dan 20–30% dari seluruh kasus glioma optik terkait dengan NF11). Setelah diagnosis NF1, pemantauan oftalmologi rutin direkomendasikan sebagai skrining glioma optik. Sebaliknya, jika glioma optik ditemukan pada anak, penting untuk memeriksa apakah kriteria diagnostik NF1 terpenuhi pada semua kasus.
Lebih sering terjadi pada wanita dewasa, dan ada kasus yang terkait dengan NF2.
Pada CT/MRI, tram-track sign (gambaran seperti rel kereta) merupakan ciri khas dan berguna untuk membedakan dari glioma saraf optik.
Neuritis Optik
Sering timbul akut dan disertai nyeri saat menggerakkan bola mata.
MRI menunjukkan peningkatan kontras pada saraf optik, namun pembengkakannya ringan. Sering membaik dengan terapi steroid.
Diagnosis banding lainnya:
Limfoma orbita (lebih sering pada dewasa)
Pseudotumor inflamasi orbita
Tumor orbital metastatik
Glioma optikus menunjukkan pembesaran seragam dan downward kinking, yang jelas dapat dibedakan dari tram-track sign meningioma sarung optikus.
QApa perbedaan antara glioma optikus dan meningioma sarung optikus?
A
Glioma optikus adalah tumor jinak (astrositoma piloid) yang sering terjadi pada anak-anak, dengan asosiasi NF1. Pada CT/MRI, ciri khasnya adalah pembesaran seragam saraf optikus dan downward kinking. Meningioma sarung optikus lebih sering pada wanita dewasa, dengan asosiasi NF2, dan pada CT/MRI menunjukkan tram-track sign (tanda seperti rel kereta: kalsifikasi atau efek kontras di sepanjang saraf optikus) yang membedakannya.
Karena tumor bersifat jinak dan sering terjadi pada anak-anak, pada prinsipnya tidak dilakukan reseksi bedah atau radioterapi jika tumor terbatas pada saraf optik intraorbital.
Prinsip dasarnya adalah observasi ketat dengan pemeriksaan pencitraan rutin (MRI: setiap 3–6 bulan).
Dahulu operasi sering dilakukan, tetapi karena risiko kebutaan ireversibel yang tinggi, kini cenderung dihindari.
Pada kasus dengan NF1, dilaporkan adanya regresi spontan, sehingga observasi ketat sangat ditekankan.
Jika terjadi penurunan visus atau pembesaran tumor yang progresif, kemoterapi kombinasi karboplatin + vinkristin (terapi CV) digunakan sebagai terapi lini pertama standar3)4).
Radioterapi dipertimbangkan pada kasus lanjut yang resisten terhadap kemoterapi. Namun, pada anak-anak, terdapat kekhawatiran mengenai risiko kanker sekunder, gangguan fungsi endokrin (iradiasi dekat hipotalamus), dan dampak pada fungsi kognitif, sehingga cenderung dihindari sebisa mungkin.
Temuan histopatologis glioma optikus adalah astrositoma pilocytic (pilocytic astrocytoma, WHO Grade I) yang bersifat jinak. Ini pada dasarnya berbeda dari glioma derajat tinggi seperti glioblastoma multiforme (WHO Grade IV).
Sel tumor menunjukkan morfologi khas dengan prosesus sel bipolar dan mengandung serat Rosenthal. Tumor berasal dari sel glial (astrosit) saraf optik, menekan dan menggantikan saraf optik dari dalam.
Gen NF1 adalah gen penekan tumor yang mengkode protein neurofibromin
neurofibromin berfungsi sebagai protein pengaktif Ras-GTPase (GAP) dan menekan sinyal Ras
Mutasi NF1 → hilangnya fungsi neurofibromin → aktivasi konstitutif jalur sinyal Ras → peningkatan sinyal MAPK → proliferasi sel glial yang tidak terkendali
Pada astrositoma piloid sporadis (tanpa NF1), gen fusi BRAF-KIAA1549 sering ditemukan. Gen fusi ini juga mengaktifkan jalur MAPK dan mendorong pertumbuhan tumor.
Beberapa kasus memiliki mutasi BRAF V600E, dan kasus dengan mutasi ini cenderung memiliki keganasan yang lebih tinggi6).
Mayoritas glioma optikus bersifat derajat rendah (low-grade) dan menunjukkan pertumbuhan lambat. Tumor membengkakkan saraf optikus dari dalam, menyebabkan saraf optikus melengkung (kinking/penekukan ke bawah) di dalam orbita. Pembengkakan seragam dan penekukan ke bawah pada MRI menjadi poin kunci diagnosis pencitraan.
Prognosis penglihatan tidak seragam; karena terdapat kasus progresif dan stabil, diperlukan evaluasi longitudinal fungsi penglihatan selain ukuran tumor. Beberapa kasus menunjukkan penurunan fungsi penglihatan meskipun temuan MRI stabil, sebaliknya pada kasus dengan NF1 dapat terjadi regresi spontan. 1, 8, 9)
Evaluasi fungsi visual: Ukur berulang kali ketajaman penglihatan, lapang pandang, penglihatan warna, dan RAPD sesuai usia
Pemantauan pencitraan: Lacak pertumbuhan kembali, perluasan ke kiasma optikum, dan perluasan intrakranial dengan MRI8)
Evaluasi endokrin: Pada kasus dengan perluasan ke hipotalamus, periksa termasuk gangguan pertumbuhan, pubertas dini, dan diabetes insipidus
Manajemen sistemik NF1: Lakukan pemantauan sistemik secara paralel termasuk lesi kulit, tumor lain, dan aspek perkembangan2, 9)
Efektivitas inhibitor MEK pada glioma derajat rendah terkait NF1 telah dilaporkan.
Dalam uji SPRINT (Fase II), dilaporkan tingkat respons objektif sebesar 66% untuk selumetinib pada neurofibroma pleksiformis progresif terkait NF17). Aplikasi pada glioma derajat rendah terkait NF1, termasuk glioma optik, sedang dipertimbangkan.
Untuk glioma derajat rendah pada anak dengan mutasi BRAF V600E, terapi kombinasi dabrafenib + trametinib telah dievaluasi dalam uji klinis6).
Pada kasus fusi BRAF-KIAA1549 positif, efektivitas inhibitor BRAF terbatas.
Dengan munculnya inhibitor MEK dan inhibitor BRAF, telah terjadi pergeseran dari kemoterapi konvensional (regimen CV) menuju pengobatan personalisasi berdasarkan profil molekuler8).
Di masa depan, pemilihan terapi berdasarkan profil mutasi genetik (fusi BRAF, BRAF V600E, mutasi NF1, dll.) berpotensi menjadi standar.
Listernick R, Charrow J, Greenwald M, Mets M. Natural history of optic pathway tumors in children with neurofibromatosis type 1: a longitudinal study. J Pediatr. 1994;125(1):63-66. doi:10.1016/s0022-3476(94)70122-9. PMID:8021787.
Ferner RE, Ferner RE, Huson SM, Thomas N, Moss C, Willshaw H, Evans DG, Upadhyaya M, Towers R, Gleeson M, Steiger C, Kirby A. Guidelines for the diagnosis and management of individuals with neurofibromatosis 1. J Med Genet. 2007;44(2):81-88. doi:10.1136/jmg.2006.045906. PMID:17105749; PMCID:PMC2598063.
Gnekow AK, Walker DA, Kandels D, Picton S, Giorgio Perilongo, Grill J, Stokland T, Sandstrom PE, Warmuth-Metz M, Pietsch T, Giangaspero F, Schmidt R, Faldum A, Kilmartin D, De Paoli A, De Salvo GL, of the Low Grade Glioma Consortium and the participating centers. A European randomised controlled trial of the addition of etoposide to standard vincristine and carboplatin induction as part of an 18-month treatment programme for childhood (≤16 years) low grade glioma - A final report. Eur J Cancer. 2017;81:206-225. doi:10.1016/j.ejca.2017.04.019. PMID:28649001; PMCID:PMC5517338.
Ater JL, Zhou T, Holmes E, Mazewski CM, Booth TN, Freyer DR, Lazarus KH, Packer RJ, Prados M, Sposto R, Vezina G, Wisoff JH, Pollack IF. Randomized study of two chemotherapy regimens for treatment of low-grade glioma in young children: a report from the Children’s Oncology Group. J Clin Oncol. 2012;30(21):2641-2647. doi:10.1200/jco.2011.36.6054. PMID:22665535; PMCID:PMC3413276.
Alvaro Lassaletta, Katrin Scheinemann, Shayna M. Zelcer, Juliette Hukin, Beverley A. Wilson, Nada Jabado, Anne Sophie Carret, Lucie Lafay-Cousin, et al. Phase II Weekly Vinblastine for Chemotherapy-Naïve Children With Progressive Low-Grade Glioma: A Canadian Pediatric Brain Tumor Consortium Study. JCO. 2016;34(29):3537-3543. doi:10.1200/jco.2016.68.1585.
Fangusaro J, Onar-Thomas A, Young Poussaint T, Wu S, Ligon AH, Lindeman N, Banerjee A, Packer RJ, Kilburn LB, Goldman S, Pollack IF, Qaddoumi I, Jakacki RI, Fisher PG, Dhall G, Baxter P, Kreissman SG, Stewart CF, Jones DTW, Pfister SM, Vezina G, Stern JS, Panigrahy A, Patay Z, Tamrazi B, Jones JY, Haque SS, Enterline DS, Cha S, Fisher MJ, Doyle LA, Smith M, Dunkel IJ, Fouladi M. Selumetinib in paediatric patients with BRAF-aberrant or neurofibromatosis type 1-associated recurrent, refractory, or progressive low-grade glioma: a multicentre, phase 2 trial. Lancet Oncol. 2019;20(7):1011-1022. doi:10.1016/s1470-2045(19)30277-3. PMID:31151904; PMCID:PMC6628202.
Anuradha Banerjee, Regina I. Jakacki, Arzu Onar-Thomas, Shengjie Wu, Theodore Nicolaides, Tina Young Poussaint, Jason Fangusaro, Joanna Phillips, et al. A phase I trial of the MEK inhibitor selumetinib (AZD6244) in pediatric patients with recurrent or refractory low-grade glioma: a Pediatric Brain Tumor Consortium (PBTC) study. Neuro-Oncology. 2017;19(8):1135-1144. doi:10.1093/neuonc/now282.
de Blank PMK, Orne-Ibekwe E, Packer R. International consensus recommendations for visual surveillance in optic pathway gliomas associated with neurofibromatosis type 1. J Neurooncol. 2020;148(3):571-578.
Listernick R, Louis DN, Packer RJ, Gutmann DH. Optic pathway gliomas in children with neurofibromatosis 1: consensus statement from the NF1 Optic Pathway Glioma Task Force. Ann Neurol. 1997;41(2):143-149. doi:10.1002/ana.410410204. PMID:9029062.
Salin teks artikel dan tempelkan ke asisten AI pilihan Anda.
Artikel disalin ke papan klip
Buka asisten AI di bawah, lalu tempelkan teks yang disalin ke kotak chat.