Intraorbital Optic Nerve–Limited Type
Most common form. Main symptoms are unilateral vision loss and proptosis.
Limited to the intraorbital optic nerve; observation is the basic approach. Spontaneous regression has been reported in cases with NF1.

Optic nerve glioma (optic pathway glioma) is a type of glioma that develops in the optic nerve. In a narrow sense, it refers to a glioma arising in the optic nerve anterior to the optic chiasm. In a broad sense, it means a glioma occurring throughout the entire optic pathway, including posterior to the optic chiasm (optic pathway glioma).
Histologically, most are benign pilocytic astrocytomas (WHO Grade I). However, some malignant cases have been reported. Approximately 70% occur in childhood, and it is a rare disease accounting for about 0.5–5% of pediatric brain tumors.
There is a strong association with neurofibromatosis type 1 (NF1, von Recklinghausen disease), and about 20–30% of optic nerve glioma cases have concurrent NF1. Conversely, optic nerve glioma is the most common orbital lesion in NF1 patients.
Intraorbital Optic Nerve–Limited Type
Most common form. Main symptoms are unilateral vision loss and proptosis.
Limited to the intraorbital optic nerve; observation is the basic approach. Spontaneous regression has been reported in cases with NF1.
Chiasmal Infiltrative Type
Type that infiltrates the optic chiasm.
It causes bilateral visual impairment and management becomes complex. It often occurs at a young age, and extension to the hypothalamus should be evaluated.
Optic pathway-hypothalamic type
A type that extends from behind the optic chiasm to the hypothalamus.
Endocrine abnormalities (such as growth disorders and precocious puberty) may be associated. Treatment requires collaboration with neurosurgery and endocrinology.
Classification by genetic background includes NF1-associated type (about 30% of all cases) and sporadic type (about 70%). Bilateral occurrence may be seen in the NF1-associated type.
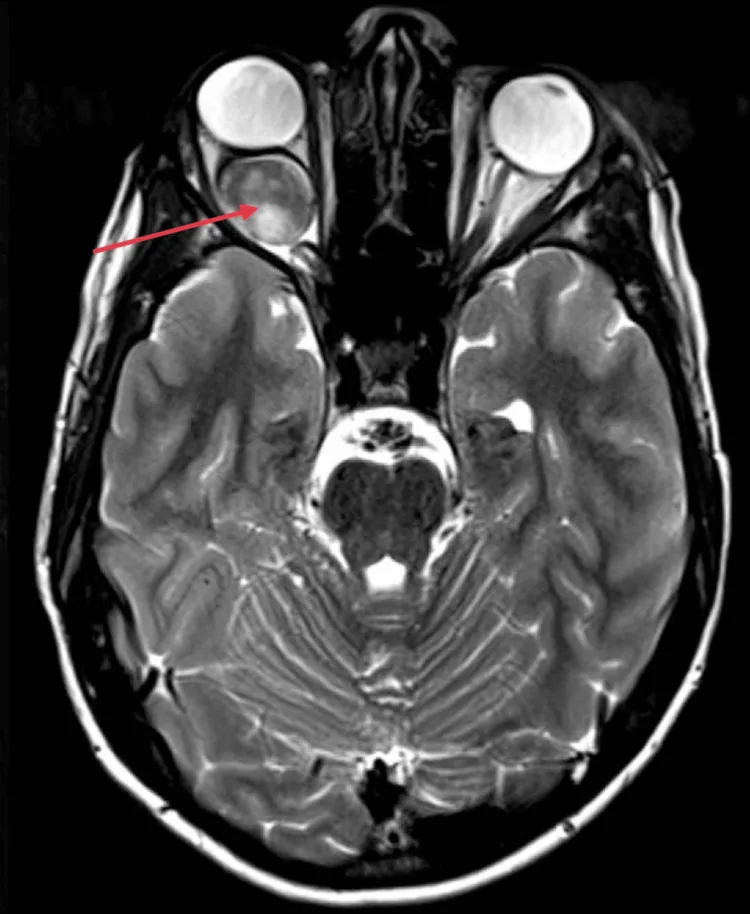
Young children do not complain of vision loss themselves. Therefore, parents or those around them often notice strabismus (especially esotropia) and bring the child for an initial ophthalmology visit.
Unilateral vision loss without strabismus is even more likely to be detected late. Optic atrophy may already be present at the initial examination.
Bilateral optic gliomas are more common in cases with early onset. They are first noticed due to abnormal eye movements or “not seeing” behaviors, and visual impairment may be severe.
Proptosis is not obvious, and there is no pain.
In optic glioma, young children often cannot perceive or report vision loss, so strabismus (especially esotropia) frequently becomes the initial symptom leading to an ophthalmology visit. In children diagnosed with strabismus, particularly unilateral strabismus, it is important to consider optic glioma and evaluate with visual acuity, fundus, and imaging studies.
Fundus examination often reveals the following findings.
In cases associated with NF1 (neurofibromatosis type 1), the following systemic findings are present.
CT findings:
MRI findings (for detailed examination):
Patients with NF1 have a significantly higher risk of optic pathway glioma, and 20–30% of all optic pathway glioma cases are associated with NF11). After an NF1 diagnosis, regular ophthalmologic follow-up is recommended for screening of optic pathway glioma. Conversely, when optic pathway glioma is found in a child, it is important to check whether the diagnostic criteria for NF1 are met in all cases.
The main items of the diagnostic criteria for NF1 (neurofibromatosis type 1) are shown below2).
| Finding | Criteria |
|---|---|
| Café-au-lait spots | ≥6 (prepubertal: longest diameter ≥5 mm; postpubertal: longest diameter ≥15 mm) |
| Neurofibromas | ≥2 of any type or ≥1 plexiform neurofibroma |
| Iris Lisch nodules | ≥2 |
| Characteristic bone lesions | Sphenoid wing dysplasia or thinning of long bone cortex |
| Optic glioma | One or more |
| Axillary or inguinal freckling | Present |
| First-degree relative | Person with a confirmed diagnosis of NF1 |
A confirmed diagnosis of NF1 requires meeting at least two of the above criteria.
MRI is superior to CT in evaluating tumor extent and is essential for detailed examination.
| Finding | Details |
|---|---|
| T1-weighted image | Shows low signal intensity |
| Gd-DTPA contrast enhancement | Uniform enlargement with strong contrast enhancement |
| Downward kinking | Characteristic in NF1-associated cases (downward bending of the optic nerve) |
| Intracranial extension assessment | Confirm extension through the optic canal into the intracranial space, and presence of tumor in the optic chiasm and hypothalamus |
Optic Nerve Sheath Meningioma
The most important disease for differential diagnosis.
It is more common in adult women and can be associated with NF2. CT/MRI shows a characteristic tram-track sign, which is useful for differentiation from optic glioma.
Optic Neuritis
Often presents with acute onset and pain on eye movement.
MRI shows contrast enhancement of the optic nerve, but swelling is mild. Often improves with steroid treatment.
Other differential diagnoses:
Optic glioma shows uniform enlargement and downward kinking, which is clearly distinguishable from the tram-track sign of optic nerve sheath meningioma.
Optic glioma is a benign tumor (pilocytic astrocytoma) common in children, often associated with NF1. CT/MRI shows uniform enlargement of the optic nerve and downward kinking. Optic nerve sheath meningioma is more common in adult women, sometimes associated with NF2, and is differentiated by the tram-track sign (calcification or contrast enhancement along the optic nerve sheath) on CT/MRI.
Because the tumor is benign and common in children, surgical resection or radiation therapy is generally not performed when the tumor is confined to the intraorbital optic nerve. The basic policy is careful observation with regular imaging (MRI every 3–6 months).
Surgery was once performed, but due to the high risk of irreversible blindness, surgical resection is now generally avoided. In NF1-associated cases, spontaneous regression has been reported, so particularly careful observation is undertaken.
When vision loss or tumor progression occurs, combination chemotherapy with carboplatin and vincristine (CV therapy) is used as standard first-line treatment3)4).
Standard CV regimen (COG A9952, etc.):
The objective response rate (partial remission + stabilization) of CV therapy is reported to be 60–80%4).
Second-line treatment options:
Considered for progressive cases resistant to chemotherapy. However, in children, there are concerns about secondary cancer risk, endocrine dysfunction (due to irradiation near the hypothalamus), and effects on cognitive function, so it tends to be avoided as much as possible.
There is currently a tendency to avoid aggressive surgical resection.
Situations where surgery may be considered:
The histopathological finding of optic glioma is benign pilocytic astrocytoma (WHO Grade I). It is fundamentally different from high-grade gliomas such as glioblastoma multiforme (WHO Grade IV).
Tumor cells show a characteristic morphology with bipolar cell processes and contain Rosenthal fibers. They arise from glial cells (astrocytes) of the optic nerve, compressing and replacing the optic nerve from within.
Optic glioma associated with NF1 (neurofibromatosis type 1) is caused by mutations in the NF1 gene (chromosome 17q11.2).
In sporadic (non-NF1-associated) pilocytic astrocytomas, the BRAF-KIAA1549 fusion gene is frequently observed. This fusion gene also activates the MAPK pathway and promotes tumor growth.
Some cases harbor the BRAF V600E mutation, and cases with this mutation tend to have higher malignancy 6).
The majority of optic gliomas are low-grade and show slow growth. The tumor enlarges the optic nerve from within, causing kinking and downward kinking of the nerve within the orbit. Uniform enlargement and downward kinking on MRI are key imaging findings.
Cases confined to the orbit:
Cases with optic chiasm/hypothalamic infiltration:
Prognosis:
Functional prognosis:
Visual prognosis is not uniform; progressive and stable cases coexist, so longitudinal assessment of visual function is necessary in addition to tumor size. Some cases show worsening of visual function despite stable MRI findings, while in NF1-associated cases, spontaneous regression may occur. 1, 8, 9)
The efficacy of MEK inhibitors for NF1-associated low-grade glioma has been reported.
In the SPRINT trial (Phase II), an objective response rate of 66% was reported for selumetinib in NF1-associated progressive low-grade glioma (plexiform neurofibroma) 7). Its application to NF1-associated low-grade gliomas, including optic pathway gliomas, is being investigated.
For pediatric low-grade gliomas with BRAF V600E mutation, combination therapy with dabrafenib plus trametinib is being evaluated in clinical trials6). In BRAF-KIAA1549 fusion-positive cases, the efficacy of BRAF inhibitors is limited.
With the advent of MEK inhibitors and BRAF inhibitors, there is a shift from conventional chemotherapy (CV therapy) to personalized treatment based on molecular profiles8). In the future, treatment selection based on genetic mutation profiles (BRAF fusion, BRAF V600E, NF1 mutation, etc.) may become standardized.
Listernick R, Charrow J, Greenwald M, Mets M. Natural history of optic pathway tumors in children with neurofibromatosis type 1: a longitudinal study. J Pediatr. 1994;125(1):63-66. doi:10.1016/s0022-3476(94)70122-9. PMID:8021787.
Ferner RE, Ferner RE, Huson SM, Thomas N, Moss C, Willshaw H, Evans DG, Upadhyaya M, Towers R, Gleeson M, Steiger C, Kirby A. Guidelines for the diagnosis and management of individuals with neurofibromatosis 1. J Med Genet. 2007;44(2):81-88. doi:10.1136/jmg.2006.045906. PMID:17105749; PMCID:PMC2598063.
Gnekow AK, Walker DA, Kandels D, Picton S, Giorgio Perilongo, Grill J, Stokland T, Sandstrom PE, Warmuth-Metz M, Pietsch T, Giangaspero F, Schmidt R, Faldum A, Kilmartin D, De Paoli A, De Salvo GL, of the Low Grade Glioma Consortium and the participating centers. A European randomised controlled trial of the addition of etoposide to standard vincristine and carboplatin induction as part of an 18-month treatment programme for childhood (≤16 years) low grade glioma - A final report. Eur J Cancer. 2017;81:206-225. doi:10.1016/j.ejca.2017.04.019. PMID:28649001; PMCID:PMC5517338.
Ater JL, Zhou T, Holmes E, Mazewski CM, Booth TN, Freyer DR, Lazarus KH, Packer RJ, Prados M, Sposto R, Vezina G, Wisoff JH, Pollack IF. Randomized study of two chemotherapy regimens for treatment of low-grade glioma in young children: a report from the Children’s Oncology Group. J Clin Oncol. 2012;30(21):2641-2647. doi:10.1200/jco.2011.36.6054. PMID:22665535; PMCID:PMC3413276.
Alvaro Lassaletta, Katrin Scheinemann, Shayna M. Zelcer, Juliette Hukin, Beverley A. Wilson, Nada Jabado, Anne Sophie Carret, Lucie Lafay-Cousin, et al. Phase II Weekly Vinblastine for Chemotherapy-Naïve Children With Progressive Low-Grade Glioma: A Canadian Pediatric Brain Tumor Consortium Study. JCO. 2016;34(29):3537-3543. doi:10.1200/jco.2016.68.1585.
Fangusaro J, Onar-Thomas A, Young Poussaint T, Wu S, Ligon AH, Lindeman N, Banerjee A, Packer RJ, Kilburn LB, Goldman S, Pollack IF, Qaddoumi I, Jakacki RI, Fisher PG, Dhall G, Baxter P, Kreissman SG, Stewart CF, Jones DTW, Pfister SM, Vezina G, Stern JS, Panigrahy A, Patay Z, Tamrazi B, Jones JY, Haque SS, Enterline DS, Cha S, Fisher MJ, Doyle LA, Smith M, Dunkel IJ, Fouladi M. Selumetinib in paediatric patients with BRAF-aberrant or neurofibromatosis type 1-associated recurrent, refractory, or progressive low-grade glioma: a multicentre, phase 2 trial. Lancet Oncol. 2019;20(7):1011-1022. doi:10.1016/s1470-2045(19)30277-3. PMID:31151904; PMCID:PMC6628202.
Anuradha Banerjee, Regina I. Jakacki, Arzu Onar-Thomas, Shengjie Wu, Theodore Nicolaides, Tina Young Poussaint, Jason Fangusaro, Joanna Phillips, et al. A phase I trial of the MEK inhibitor selumetinib (AZD6244) in pediatric patients with recurrent or refractory low-grade glioma: a Pediatric Brain Tumor Consortium (PBTC) study. Neuro-Oncology. 2017;19(8):1135-1144. doi:10.1093/neuonc/now282.
de Blank PMK, Orne-Ibekwe E, Packer R. International consensus recommendations for visual surveillance in optic pathway gliomas associated with neurofibromatosis type 1. J Neurooncol. 2020;148(3):571-578.
Listernick R, Louis DN, Packer RJ, Gutmann DH. Optic pathway gliomas in children with neurofibromatosis 1: consensus statement from the NF1 Optic Pathway Glioma Task Force. Ann Neurol. 1997;41(2):143-149. doi:10.1002/ana.410410204. PMID:9029062.