ชนิดจำกัดเฉพาะเส้นประสาทตาในเบ้าตา
รูปแบบที่พบบ่อยที่สุด อาการหลักคือการมองเห็นลดลงข้างเดียวและตาโปน
จำกัดเฉพาะเส้นประสาทตาในเบ้าตา โดยทั่วไปจะสังเกตอาการ ในผู้ป่วยร่วมกับ NF1 มีรายงานการหดเล็กลงเอง

เนื้องอกไกลโอมาของเส้นประสาทตา (optic nerve glioma / optic pathway glioma) เป็นเนื้องอกไกลโอมาชนิดหนึ่งที่เกิดขึ้นในเส้นประสาทตา ในความหมายแคบหมายถึงเนื้องอกไกลโอมาที่เกิดในเส้นประสาทตาส่วนหน้าของออปติกไคแอสมาส์ ในความหมายกว้างหมายถึงเนื้องอกไกลโอมาที่เกิดในวิถีประสาทตาทั้งหมดรวมถึงส่วนหลังของออปติกไคแอสมาส์ (optic pathway glioma)
ในทางจุลพยาธิวิทยา ส่วนใหญ่เป็นพิโลไซติกแอสโตรไซโตมา (pilocytic astrocytoma, WHO Grade I) ซึ่งเป็นเนื้องอกชนิดไม่ร้ายแรง อย่างไรก็ตาม มีรายงานบางส่วนที่เป็นชนิดร้ายแรง ประมาณ 70% เกิดในวัยเด็ก และเป็นโรคที่พบได้ยาก คิดเป็นประมาณ 0.5-5% ของเนื้องอกสมองในเด็ก
มีความสัมพันธ์อย่างมากกับโรคนิวโรไฟโบรมาทอซิสชนิดที่ 1 (NF1, von Recklinghausen disease) โดยประมาณ 20-30% ของผู้ป่วยเนื้องอกไกลโอมาของเส้นประสาทตามี NF1 ร่วมด้วย ในทางกลับกัน เนื้องอกไกลโอมาของเส้นประสาทตาเป็นรอยโรคในเบ้าตาที่พบบ่อยที่สุดในผู้ป่วย NF1
ชนิดจำกัดเฉพาะเส้นประสาทตาในเบ้าตา
รูปแบบที่พบบ่อยที่สุด อาการหลักคือการมองเห็นลดลงข้างเดียวและตาโปน
จำกัดเฉพาะเส้นประสาทตาในเบ้าตา โดยทั่วไปจะสังเกตอาการ ในผู้ป่วยร่วมกับ NF1 มีรายงานการหดเล็กลงเอง
ชนิดลุกลามถึงออปติกไคแอสมาหรือจุดไขว้ประสาทตา
ชนิดที่ลุกลามถึงออปติกไคแอสมาหรือจุดไขว้ประสาทตา
ทำให้เกิดความบกพร่องทางการมองเห็นทั้งสองข้างและการจัดการมีความซับซ้อน มักเกิดในเด็กเล็กและต้องประเมินการลุกลามไปยังไฮโปทาลามัส
ชนิดเส้นทางประสาทตา-ไฮโปทาลามัส
ชนิดที่ลุกลามจากด้านหลังของออปติกไคแอสมาสไปยังไฮโปทาลามัส
อาจมีภาวะผิดปกติของต่อมไร้ท่อร่วมด้วย (เช่น ปัญหาการเจริญเติบโต ภาวะ puberty ก่อนวัย) ในการรักษาจำเป็นต้องทำงานร่วมกันระหว่างศัลยกรรมประสาทและอายุรศาสตร์ต่อมไร้ท่อ
การจำแนกตามภูมิหลังทางพันธุกรรมมี 2 ชนิด: ชนิดที่เกิดร่วมกับ NF1 (ประมาณ 30% ของทั้งหมด) และชนิดประปราย (ประมาณ 70%) ในชนิดที่เกิดร่วมกับ NF1 อาจพบการเกิดทั้งสองข้าง
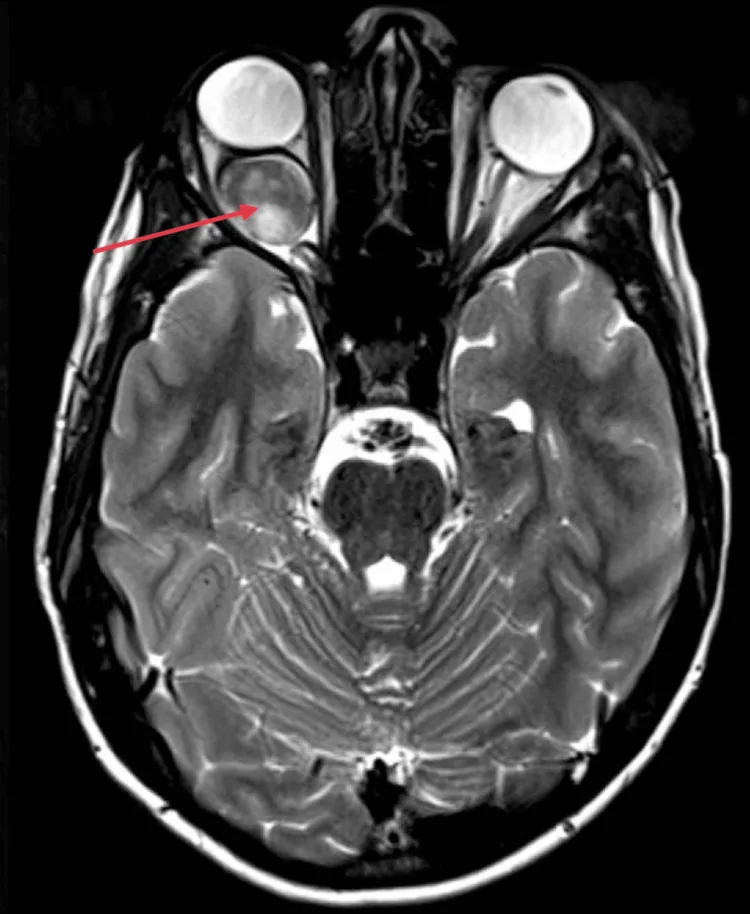
เด็กเล็กไม่สามารถบอกได้เองว่าสายตาลดลง ดังนั้นผู้ปกครองหรือคนรอบข้างมักสังเกตเห็นตาเหล่ (โดยเฉพาะตาเหล่เข้า) และพามาพบจักษุแพทย์เป็นครั้งแรก
การมองเห็นลดลงข้างเดียวโดยไม่มีตาเหล่มักถูกตรวจพบช้ากว่า บางครั้งเมื่อมาพบแพทย์ครั้งแรกก็พบว่ามีการฝ่อของเส้นประสาทตาแล้ว
เนื้องอกเส้นประสาทตาชนิดสองตามักพบในผู้ป่วยที่อายุน้อย มักสังเกตได้จากความผิดปกติของการมองหรือพฤติกรรมที่ “มองไม่เห็น” และอาจมีภาวะสายตาบกพร่องอย่างรุนแรง
ตาโปนไม่ชัดเจน และไม่มีอาการปวด
ในเนื้องอกแก้วนำแสง เด็กเล็กมักไม่สามารถรับรู้หรือบอกอาการตามัวได้ ดังนั้นตาเหล่ (โดยเฉพาะตาเหล่เข้า) จึงเป็นอาการเริ่มแรกที่พาไปพบจักษุแพทย์ได้บ่อย ในเด็กที่ได้รับการวินิจฉัยว่าเป็นตาเหล่ โดยเฉพาะตาเหล่ข้างเดียว ควรพิจารณาตรวจเพิ่มเติมรวมถึงการตรวจวัดสายตา ตรวจอวัยวะภายในตา และการถ่ายภาพเพื่อหาเนื้องอกแก้วนำแสง
การตรวจอวัยวะภายในตามักพบผลดังต่อไปนี้
ในผู้ป่วยร่วมกับ NF1 (neurofibromatosis type 1) จะมีอาการทางระบบดังต่อไปนี้
ภาพ CT:
ผลMRI (การตรวจละเอียด):
ผู้ป่วย NF1 มีความเสี่ยงสูงมากต่อการเกิด optic pathway glioma โดย 20-30% ของผู้ป่วย optic pathway glioma ทั้งหมดมี NF1 ร่วมด้วย1) หลังการวินิจฉัย NF1 แนะนำให้ติดตามทางจักษุวิทยาเป็นระยะเพื่อคัดกรอง optic pathway glioma ในทางกลับกัน หากพบ optic pathway glioma ในเด็ก ควรตรวจสอบว่าผู้ป่วยรายนั้นเข้าเกณฑ์การวินิจฉัย NF1 หรือไม่ในทุกราย
เกณฑ์หลักในการวินิจฉัย NF1 (neurofibromatosis type 1) แสดงดังต่อไปนี้2)
| ลักษณะที่พบ | เกณฑ์ |
|---|---|
| จุด café-au-lait | ≥6 จุด (เด็ก: เส้นผ่านศูนย์กลาง ≥5 มม., วัยรุ่นขึ้นไป: เส้นผ่านศูนย์กลาง ≥15 มม.) |
| เนื้องอกเส้นประสาท (neurofibroma) | ≥2 ก้อน (ชนิดใดก็ได้) หรือ neurofibroma แบบ plexiform อย่างน้อย 1 ก้อน |
| ก้อน Lisch ที่ม่านตา | ≥2 ก้อน |
| รอยโรคกระดูกที่มีลักษณะเฉพาะ | ความผิดปกติของปีกกระดูกสฟีนอยด์หรือการบางของเยื่อหุ้มกระดูกยาว |
| เนื้องอกไกลโอมาของเส้นประสาทตา | 1 รอยโรคขึ้นไป |
| กระที่รักแร้หรือขาหนีบ | มีอยู่ |
| สมาชิกในครอบครัวลำดับที่หนึ่ง | ผู้ที่ได้รับการวินิจฉัยแน่ชัดว่าเป็น NF1 |
การวินิจฉัยแน่ชัดของ NF1 จำเป็นต้องมีเกณฑ์ข้างต้นอย่างน้อย 2 ข้อ
MRI เหนือกว่า CT ในการประเมินขอบเขตการแพร่กระจายของเนื้องอก และเป็นการตรวจที่จำเป็นสำหรับการตรวจอย่างละเอียด
| ผลการตรวจ | รายละเอียด |
|---|---|
| T1-weighted image | แสดงสัญญาณต่ำ |
| การเพิ่มความเข้มของ Gd-DTPA | แสดงการโตขึ้นอย่างสม่ำเสมอและการเพิ่มความเข้มที่รุนแรง |
| downward kinking | ลักษณะเฉพาะในผู้ป่วยร่วมกับ NF1 (การโค้งงอลงของเส้นประสาทตา) |
| การประเมินการลุกลามเข้าสู่กะโหลกศีรษะ | ยืนยันการลุกลามจากภายในคลองประสาทตาเข้าสู่กะโหลกศีรษะ การมีเนื้องอกที่ออปติกไคแอสมและไฮโปทาลามัส |
เยื่อหุ้มประสาทตาเนื้องอกชนิดเมนินจิโอมา
โรคที่สำคัญที่สุดที่ต้องแยก
พบบ่อยในหญิงวัยผู้ใหญ่ และอาจพบร่วมกับ NF2 ลักษณะเฉพาะใน CT/MRI คือ tram-track sign (ลักษณะคล้ายรางรถไฟ) ซึ่งมีประโยชน์ในการแยกจากเนื้องอกแก้วนำแสงชนิดไกลโอมา
เส้นประสาทตาอักเสบ
มักเริ่มมีอาการเฉียบพลันและมีอาการปวดเมื่อขยับลูกตา
MRI พบการเพิ่มความเข้มของเส้นประสาทตา แต่มีการบวมเล็กน้อย มักตอบสนองต่อการรักษาด้วยสเตียรอยด์
โรคอื่นที่ต้องแยกวินิจฉัย:
Optic glioma แสดงลักษณะ การขยายตัวที่สม่ำเสมอและ downward kinking ซึ่งแตกต่างจาก tram-track sign ของ meningioma ของปลอกประสาทตาได้อย่างชัดเจน
Optic glioma เป็นเนื้องอกชนิดไม่ร้าย (pilocytic astrocytoma) ที่พบบ่อยในเด็ก และอาจพบร่วมกับ NF1 การตรวจ CT/MRI พบลักษณะเฉพาะคือการขยายตัวที่สม่ำเสมอของเส้นประสาทตาและ downward kinking ส่วน meningioma ของปลอกประสาทตาพบมากในหญิงวัยผู้ใหญ่ อาจพบร่วมกับ NF2 และการตรวจ CT/MRI พบ tram-track sign (ลักษณะคล้ายรางรถไฟ: การกลายเป็นปูนหรือการเพิ่มความเข้มของสารทึบรังสีตามแนวรอบเส้นประสาทตา) ซึ่งใช้ในการแยกโรค
เนื่องจากเนื้องอกเป็นชนิดไม่ร้ายแรงและพบได้บ่อยในเด็ก หากเนื้องอกจำกัดอยู่เฉพาะในเส้นประสาทตาในเบ้าตา โดยหลักการแล้วจะไม่ทำการผ่าตัดหรือฉายรังสี แนวทางหลักคือการติดตามสังเกตอาการอย่างระมัดระวัง โดยเน้นการตรวจภาพด้วยคลื่นแม่เหล็กไฟฟ้า (MRI) ทุก 3-6 เดือน
ในอดีตเคยมีการผ่าตัด แต่เนื่องจากมีความเสี่ยงสูงที่จะทำให้ตาบอดถาวร ปัจจุบันจึงมีแนวโน้มหลีกเลี่ยงการผ่าตัด ในผู้ป่วยที่ร่วมกับ NF1 มีรายงานการหดเล็กลงเองของเนื้องอก ดังนั้นจึงต้องติดตามสังเกตอาการอย่างระมัดระวังเป็นพิเศษ
ในกรณีที่การมองเห็นลดลงหรือเนื้องอกมีขนาดใหญ่ขึ้น การใช้เคมีบำบัดร่วมระหว่าง คาร์โบพลาตินและวินคริสทีน (CV therapy) ถือเป็นการรักษามาตรฐานบรรทัดแรก3)4)
สูตรมาตรฐานของ CV therapy (COG A9952 ฯลฯ):
อัตราการตอบสนองตามวัตถุประสงค์ (การทุเลาบางส่วน + การคงที่) ของ CV therapy รายงานว่าอยู่ที่ 60–80% 4)
ทางเลือกในการรักษาแนวที่สอง:
พิจารณาในกรณีที่โรคลุกลามและดื้อต่อเคมีบำบัด อย่างไรก็ตาม ในเด็กมีความกังวลเกี่ยวกับความเสี่ยงของมะเร็งทุติยภูมิ ความผิดปกติของต่อมไร้ท่อ (จากการฉายรังสีบริเวณใกล้ไฮโปทาลามัส) และผลกระทบต่อการรู้คิด จึงมีแนวโน้มที่จะหลีกเลี่ยงให้มากที่สุด
ปัจจุบันมีแนวโน้มที่จะหลีกเลี่ยงการผ่าตัดเอาออกอย่างรุนแรง
สถานการณ์ที่พิจารณาให้ทำการผ่าตัด:
ลักษณะทางจุลพยาธิวิทยาของออปติกไกลโอมาคือ พิโลไซติกแอสโตรไซโตมา (pilocytic astrocytoma, WHO Grade I) ซึ่งเป็นชนิดไม่ร้ายแรง แตกต่างโดยพื้นฐานจากไกลโอมาชนิดร้ายแรง เช่น ไกลโอบลาสโตมา มัลติฟอร์ม (glioblastoma multiforme, WHO Grade IV)
เซลล์เนื้องอกมีลักษณะเฉพาะคือมีส่วนยื่นของเซลล์แบบสองขั้ว และมีเส้นใยโรเซนธัล (Rosenthal fibers) เกิดจากเซลล์เกลีย (แอสโตรไซต์) ของเส้นประสาทตา และกดทับหรือแทนที่เส้นประสาทตาจากภายใน
ออปติกไกลโอมาที่เกี่ยวข้องกับ NF1 (นิวโรไฟโบรมาติซิสชนิดที่ 1) เกิดจากการกลายพันธุ์ของ ยีน NF1 (โครโมโซม 17q11.2)
ใน pilocytic astrocytoma ที่เกิดขึ้นเอง (ไม่เกี่ยวข้องกับ NF1) มักพบ ยีนฟิวชัน BRAF-KIAA1549 บ่อยครั้ง ยีนฟิวชันนี้ยังกระตุ้นวิถี MAPK และส่งเสริมการเจริญเติบโตของเนื้องอก
ในบางกรณีมีการกลายพันธุ์ BRAF V600E ซึ่งผู้ป่วยที่มีการกลายพันธุ์นี้มักมีระดับความร้ายแรงสูงกว่า6)
เนื้องอกแก้วนำแสงส่วนใหญ่เป็น เกรดต่ำ (low-grade) และมีการเจริญเติบโตช้า เนื้องอกจะทำให้เส้นประสาทตาบวมจากภายใน และทำให้เส้นประสาทตางอ (kinking/downward kinking) ในเบ้าตา การบวมอย่างสม่ำเสมอและการโค้งลงด้านล่างใน MRI เป็นจุดสำคัญในการวินิจฉัยด้วยภาพ
กรณีที่จำกัดอยู่ในเบ้าตา:
กรณีที่มีการลุกลามไปยังออปติกไคแอสมและไฮโปทาลามัส:
การพยากรณ์โรคในระยะยาว:
การพยากรณ์โรคด้านการทำงาน:
การพยากรณ์โรคด้านการมองเห็นไม่สม่ำเสมอ โดยมีทั้งกรณีที่อาการแย่ลงและคงที่ ดังนั้นจึงจำเป็นต้องประเมินการมองเห็นตามยาว ไม่ใช่แค่ขนาดของเนื้องอกเท่านั้น มีบางกรณีที่การมองเห็นแย่ลงแม้ผล MRI จะคงที่ ในทางกลับกัน ในผู้ป่วยที่ร่วมกับ NF1 เนื้องอกอาจหดเล็กลงเองได้ 1, 8, 9)
มีการรายงานประสิทธิภาพของสารยับยั้ง MEK ต่อเนื้องอกเกลียระดับต่ำที่เกี่ยวข้องกับ NF1
ในการทดลอง SPRINT (ระยะที่ 2) รายงานอัตราการตอบสนองตามวัตถุประสงค์ 66% ของเซลูเมทินิบต่อเนื้องอกเกลียระดับต่ำที่ลุกลาม (neurofibroma แบบ plexiform) ที่เกี่ยวข้องกับ NF1 7) กำลังมีการศึกษาการประยุกต์ใช้กับเนื้องอกเกลียระดับต่ำที่เกี่ยวข้องกับ NF1 รวมถึง optic glioma
สำหรับเด็กที่มีเนื้องอกเกลียระดับต่ำที่มีการกลายพันธุ์ BRAF V600E การรักษาแบบผสมผสานด้วย ดาบราเฟนิบ + ทราเมทินิบ กำลังถูกประเมินในการทดลองทางคลินิก6) ในกรณีที่ผลบวกต่อ BRAF-KIAA1549 fusion ประสิทธิภาพของยา BRAF inhibitor มีจำกัด
การเกิดขึ้นของ MEK inhibitor และ BRAF inhibitor ทำให้เกิดการเปลี่ยนแปลงจากการรักษาด้วยเคมีบำบัดแบบดั้งเดิม (CV therapy) ไปสู่ การรักษาเฉพาะบุคคลตามโปรไฟล์โมเลกุล8) ในอนาคต การเลือกการรักษาตามโปรไฟล์การกลายพันธุ์ของยีน (BRAF fusion, BRAF V600E, NF1 mutation ฯลฯ) อาจกลายเป็นมาตรฐาน
Listernick R, Charrow J, Greenwald M, Mets M. Natural history of optic pathway tumors in children with neurofibromatosis type 1: a longitudinal study. J Pediatr. 1994;125(1):63-66. doi:10.1016/s0022-3476(94)70122-9. PMID:8021787.
Ferner RE, Ferner RE, Huson SM, Thomas N, Moss C, Willshaw H, Evans DG, Upadhyaya M, Towers R, Gleeson M, Steiger C, Kirby A. Guidelines for the diagnosis and management of individuals with neurofibromatosis 1. J Med Genet. 2007;44(2):81-88. doi:10.1136/jmg.2006.045906. PMID:17105749; PMCID:PMC2598063.
Gnekow AK, Walker DA, Kandels D, Picton S, Giorgio Perilongo, Grill J, Stokland T, Sandstrom PE, Warmuth-Metz M, Pietsch T, Giangaspero F, Schmidt R, Faldum A, Kilmartin D, De Paoli A, De Salvo GL, of the Low Grade Glioma Consortium and the participating centers. A European randomised controlled trial of the addition of etoposide to standard vincristine and carboplatin induction as part of an 18-month treatment programme for childhood (≤16 years) low grade glioma - A final report. Eur J Cancer. 2017;81:206-225. doi:10.1016/j.ejca.2017.04.019. PMID:28649001; PMCID:PMC5517338.
Ater JL, Zhou T, Holmes E, Mazewski CM, Booth TN, Freyer DR, Lazarus KH, Packer RJ, Prados M, Sposto R, Vezina G, Wisoff JH, Pollack IF. Randomized study of two chemotherapy regimens for treatment of low-grade glioma in young children: a report from the Children’s Oncology Group. J Clin Oncol. 2012;30(21):2641-2647. doi:10.1200/jco.2011.36.6054. PMID:22665535; PMCID:PMC3413276.
Alvaro Lassaletta, Katrin Scheinemann, Shayna M. Zelcer, Juliette Hukin, Beverley A. Wilson, Nada Jabado, Anne Sophie Carret, Lucie Lafay-Cousin, et al. Phase II Weekly Vinblastine for Chemotherapy-Naïve Children With Progressive Low-Grade Glioma: A Canadian Pediatric Brain Tumor Consortium Study. JCO. 2016;34(29):3537-3543. doi:10.1200/jco.2016.68.1585.
Fangusaro J, Onar-Thomas A, Young Poussaint T, Wu S, Ligon AH, Lindeman N, Banerjee A, Packer RJ, Kilburn LB, Goldman S, Pollack IF, Qaddoumi I, Jakacki RI, Fisher PG, Dhall G, Baxter P, Kreissman SG, Stewart CF, Jones DTW, Pfister SM, Vezina G, Stern JS, Panigrahy A, Patay Z, Tamrazi B, Jones JY, Haque SS, Enterline DS, Cha S, Fisher MJ, Doyle LA, Smith M, Dunkel IJ, Fouladi M. Selumetinib in paediatric patients with BRAF-aberrant or neurofibromatosis type 1-associated recurrent, refractory, or progressive low-grade glioma: a multicentre, phase 2 trial. Lancet Oncol. 2019;20(7):1011-1022. doi:10.1016/s1470-2045(19)30277-3. PMID:31151904; PMCID:PMC6628202.
Anuradha Banerjee, Regina I. Jakacki, Arzu Onar-Thomas, Shengjie Wu, Theodore Nicolaides, Tina Young Poussaint, Jason Fangusaro, Joanna Phillips, et al. A phase I trial of the MEK inhibitor selumetinib (AZD6244) in pediatric patients with recurrent or refractory low-grade glioma: a Pediatric Brain Tumor Consortium (PBTC) study. Neuro-Oncology. 2017;19(8):1135-1144. doi:10.1093/neuonc/now282.
de Blank PMK, Orne-Ibekwe E, Packer R. International consensus recommendations for visual surveillance in optic pathway gliomas associated with neurofibromatosis type 1. J Neurooncol. 2020;148(3):571-578.
Listernick R, Louis DN, Packer RJ, Gutmann DH. Optic pathway gliomas in children with neurofibromatosis 1: consensus statement from the NF1 Optic Pathway Glioma Task Force. Ann Neurol. 1997;41(2):143-149. doi:10.1002/ana.410410204. PMID:9029062.