Optik sinir gliomu (optic nerve glioma / optic pathway glioma), optik sinirde oluşan bir glioma türüdür. Dar anlamda, optik kiazmanın önündeki optik sinirde oluşan gliomu ifade eder. Geniş anlamda, optik kiazmanın arkasını da içeren tüm optik yol boyunca oluşan gliomu (optic pathway glioma) ifade eder.
Histolojik olarak, çoğu iyi huylu pilositik astrositomdur (pilocytic astrocytoma, WHO Grade I). Ancak bazı kötü huylu vakalar da bildirilmiştir. Yaklaşık %70’i çocukluk çağında ortaya çıkar ve çocukluk çağı beyin tümörlerinin yaklaşık %0.5-5’ini oluşturan nadir bir hastalıktır.
Nörofibromatozis tip 1 (NF1, von Recklinghausen hastalığı) ile güçlü bir ilişkisi vardır ve optik sinir gliomu vakalarının yaklaşık %20-30’unda NF1 birlikteliği görülür. Tersine, NF1 hastalarında en sık görülen orbital lezyon optik sinir gliomudur.
En sık görülen gelişim şekli. Tek taraflı görme azalması ve göz küresinde öne doğru çıkıntı (proptozis) ana belirtilerdir.
Göz çukurundaki optik sinirle sınırlıdır ve temel yaklaşım izlemdir.
NF1 birlikteliğinde kendiliğinden küçülme bildirilmiştir.
Kiazma İnvazyonlu Tip
Optik kiazmaya kadar yayılım gösteren tiptir.
Her iki gözde görme bozukluğuna neden olur ve yönetimi karmaşıktır. Genellikle erken yaşta ortaya çıkar ve hipotalamusa yayılımı değerlendirilmelidir.
Optik yol-hipotalamik tip
Kiazmanın arkasından hipotalamusa kadar uzanan tiptir.
Endokrin anormallikler (büyüme bozukluğu, erken ergenlik vb.) eşlik edebilir. Tedavide nöroşirürji ve endokrinoloji ile iş birliği gereklidir.
Genetik arka plana göre sınıflandırmada NF1 ilişkili tip (yaklaşık %30) ve sporadik tip (yaklaşık %70) bulunur. NF1 ilişkili tipte bilateral oluşum da görülebilir.
Kotecha MR, et al. Idiopathic Optic Nerve Glioma: A Case Report. Cureus. 2024. Figure 2. PMCID: PMC11744336. License: CC BY.
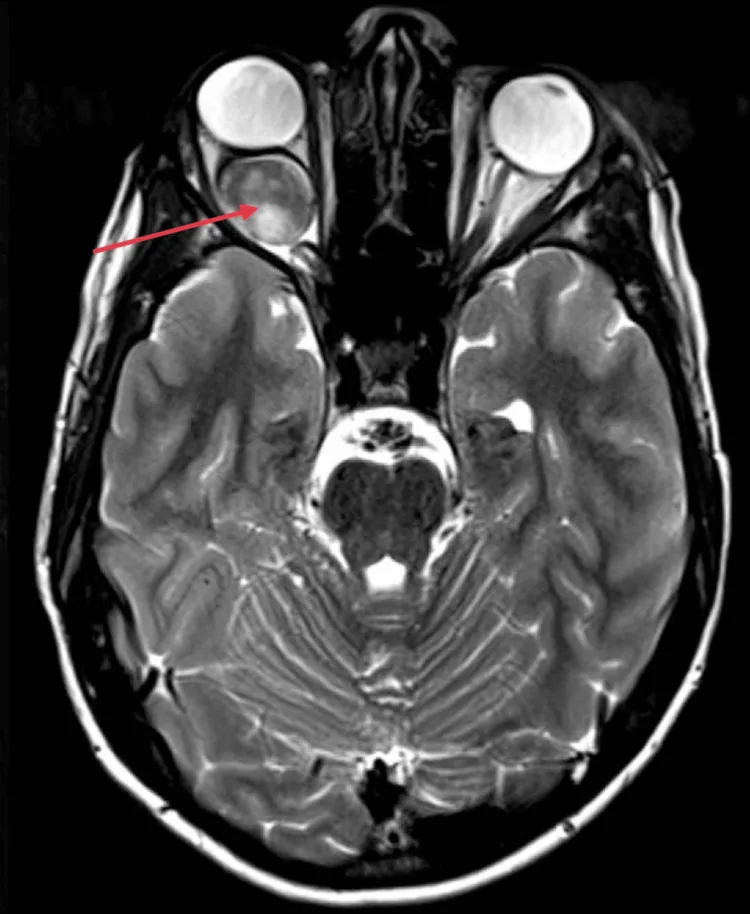
Aksiyel MR’da sağ orbital optik sinir iğ şeklinde genişlemiş, homojen ve güçlü kontrast tutulumu (kırmızı ok) göstermekte olup, göz küresi arkasındaki kitle etkisine bağlı proptozis izlenmektedir. Bu görüntü, metnin “2. Başlıca belirtiler ve klinik bulgular” bölümünde ele alınan MR bulgularına (optik sinirin homojen genişlemesi, downward kinking) karşılık gelmektedir.
Küçük çocuklar görme azalmasından kendiliğinden şikayet etmezler. Bu nedenle ebeveynler veya çevredekiler şaşılık (özellikle içe şaşılık) fark eder ve çocuk ilk kez göz doktoruna götürülür.
Şaşılık olmayan tek taraflı görme azalması daha da geç fark edilir. İlk muayenede zaten optik sinir atrofisi gelişmiş olabilir.
İki taraflı optik sinir gliomu daha çok küçük yaşta başlayan vakalarda görülür. Göz hareketlerinde anormallik veya “görmeme davranışı” ile fark edilir ve görme kaybı ciddi olabilir.
Göz küresinde belirgin bir dışarı çıkma yoktur ve ağrı da olmaz.
QÇocuğuma şaşılık teşhisi konulursa, optik gliom olasılığı var mıdır?
A
Optik gliomda, küçük yaştaki çocuklar görme azalmasını fark edip ifade edemediklerinden, şaşılık (özellikle içe şaşılık) ilk belirti olarak göz muayenesine başvurma nedeni olabilir. Şaşılık tanısı konan çocuklarda, özellikle tek taraflı şaşılıkta, optik gliom akılda tutularak görme keskinliği, fundus ve görüntüleme testlerini içeren ileri inceleme düşünülmelidir.
NF1’in orbital lezyonlarında en sık optik gliom görülür
QNF1 (nörofibromatozis tip 1) varlığında optik gliom gelişme riski artar mı?
A
NF1 hastalarında optik gliom riski belirgin şekilde yüksektir ve tüm optik gliom vakalarının %20-30’u NF1 ile ilişkilidir1). NF1 tanısı sonrası optik gliom taraması için düzenli oftalmolojik takip önerilir. Tersine, bir çocukta optik gliom saptandığında, NF1 tanı kriterlerinin karşılanıp karşılanmadığının tüm vakalarda kontrol edilmesi önemlidir.
Yetişkin kadınlarda daha sık görülür ve NF2 ile ilişkili olabilir.
BT/MRG’de tram-track sign (tramvay yolu görünümü) karakteristiktir ve optik gliomdan ayırt etmede yararlıdır.
Optik Nörit
Sıklıkla akut başlangıçlı ve göz hareketlerinde ağrı ile birlikte görülür.
MRG’de optik sinirde kontrast tutulumu görülür, ancak şişlik hafiftir. Steroid tedavisi ile sıklıkla düzelir.
Optik gliomuniform kalınlaşma ve aşağı doğru kıvrılma (downward kinking) gösterir ve optik sinir kılıfı menenjiyomunun tram-track bulgusundan açıkça ayırt edilebilir.
QOptik gliom ile optik sinir kılıfı menenjiyomu arasındaki fark nedir?
A
Optik gliom çocuklarda sık görülen benign bir tümördür (pilositik astrositom) ve NF1 birlikteliği olabilir. BT/MRG’de optik sinirin uniform kalınlaşması ve aşağı doğru kıvrılması (downward kinking) karakteristiktir. Optik sinir kılıfı menenjiyomu ise erişkin kadınlarda daha sık görülür, NF2 birlikteliği olabilir ve BT/MRG’de tram-track bulgusu (optik sinir çevresinde kalsifikasyon veya kontrast tutulumu) ile ayırt edilir.
Tümör iyi huyludur ve çocuklarda sık görülür; bu nedenle göz içi optik sinirle sınırlı olduğunda cerrahi rezeksiyon veya radyoterapi prensip olarak uygulanmaz. Düzenli görüntüleme (MRI: 3-6 ayda bir) ile dikkatli gözlem temel yaklaşımdır.
Geçmişte cerrahi uygulanıyordu ancak geri dönüşümsüz körlük riski yüksek olduğundan günümüzde cerrahi rezeksiyondan kaçınılma eğilimi vardır. NF1 birlikteliğinde spontan regresyon bildirildiğinden özellikle dikkatli gözlem yapılır.
Görme azalması veya tümör büyümesi ilerlediğinde, karboplatin + vinkristin (KV tedavisi) kombinasyon kemoterapisi standart birinci basamak tedavi olarak kullanılır3)4).
CV tedavisinin standart rejimi (COG A9952 vb.):
Vinkristin: 1.5 mg/m² IV, haftada 1 kez × 10 hafta
Kemoterapiye dirençli ilerlemiş vakalarda düşünülür. Ancak çocuklarda ikincil kanser riski, endokrin bozukluklar (hipotalamus yakınına ışınlama) ve bilişsel işlevler üzerindeki etkiler nedeniyle mümkün olduğunca kaçınılma eğilimindedir.
Optik gliomun histopatolojik bulgusu, benign pilositik astrositom (pilocytic astrocytoma, WHO Grade I)‘dur. Glioblastoma multiforme (WHO Grade IV) gibi yüksek dereceli gliomlardan temelde farklıdır.
Tümör hücreleri, bipolar hücre uzantılarına sahip karakteristik bir morfoloji gösterir ve Rosenthal lifleri içerir. Optik sinirin glial hücrelerinden (astrositler) kaynaklanır ve optik siniri içeriden sıkıştırıp yer değiştirir.
Sporadik (NF1 ile ilişkili olmayan) pilositik astrositomlarda BRAF-KIAA1549 füzyon geni sık görülür. Bu füzyon geni de MAPK yolunu aktive ederek tümör büyümesini teşvik eder.
Bazı vakalarda BRAF V600E mutasyonu bulunur ve bu mutasyona sahip vakalar daha yüksek malignite eğilimindedir6).
Optik gliomların çoğu düşük dereceli (low-grade) olup yavaş büyüme gösterir. Tümör optik siniri içeriden genişleterek yörünge içinde sinirin bükülmesine (kinking, aşağı doğru bükülme) neden olur. MRG’de homojen genişleme ve aşağı doğru bükülme görüntülemede anahtar noktalardır.
Görme prognozu tek tip değildir; ilerleyen ve stabil vakalar bir arada bulunduğundan, sadece tümör boyutuna değil, aynı zamanda görme fonksiyonunun boylamsal değerlendirmesine de ihtiyaç vardır. MRG bulguları stabil olsa bile görme fonksiyonunun kötüleştiği vakalar olabilir; tersine, NF1 ile ilişkili vakalarda spontan gerileme görülebilir. 1, 8, 9)
Görsel fonksiyon değerlendirmesi: Görme keskinliği, görme alanı, renk görme ve RAPD yaşa uygun şekilde tekrarlanarak ölçülür
Görüntüleme takibi: MRI ile yeniden büyüme, kiazma yayılımı ve intrakraniyal yayılım izlenir8)
Endokrin değerlendirme: Hipotalamik yayılım vakalarında büyüme bozukluğu, erken ergenlik ve diabetes insipidus dahil olmak üzere kontrol edilir
NF1 sistemik yönetimi: Deri lezyonları, diğer tümörler ve gelişimsel yönler dahil olmak üzere sistemik takip eş zamanlı yürütülür2, 9)
NF1 ile ilişkili düşük dereceli gliomda MEK inhibitörünün etkinliği rapor edilmiştir.
SPRINT çalışmasında (Faz II), NF1 ile ilişkili ilerleyici düşük dereceli gliom (pleksiform nörofibrom) için selumetinibin objektif yanıt oranı %66 olarak bildirilmiştir7).
Optik gliom dahil NF1 ile ilişkili düşük dereceli gliomda uygulanması araştırılmaktadır.
MEK inhibitörleri ve BRAF inhibitörlerinin ortaya çıkışıyla, geleneksel kemoterapiden (CV tedavisi) moleküler profile dayalı kişiselleştirilmiş tedaviye geçiş hızlanmaktadır8). Gelecekte, genetik mutasyon profiline (BRAF füzyonu, BRAF V600E, NF1 mutasyonu vb.) dayalı tedavi seçiminin standart hale gelmesi mümkündür.
Listernick R, Charrow J, Greenwald M, Mets M. Natural history of optic pathway tumors in children with neurofibromatosis type 1: a longitudinal study. J Pediatr. 1994;125(1):63-66. doi:10.1016/s0022-3476(94)70122-9. PMID:8021787.
Ferner RE, Ferner RE, Huson SM, Thomas N, Moss C, Willshaw H, Evans DG, Upadhyaya M, Towers R, Gleeson M, Steiger C, Kirby A. Guidelines for the diagnosis and management of individuals with neurofibromatosis 1. J Med Genet. 2007;44(2):81-88. doi:10.1136/jmg.2006.045906. PMID:17105749; PMCID:PMC2598063.
Gnekow AK, Walker DA, Kandels D, Picton S, Giorgio Perilongo, Grill J, Stokland T, Sandstrom PE, Warmuth-Metz M, Pietsch T, Giangaspero F, Schmidt R, Faldum A, Kilmartin D, De Paoli A, De Salvo GL, of the Low Grade Glioma Consortium and the participating centers. A European randomised controlled trial of the addition of etoposide to standard vincristine and carboplatin induction as part of an 18-month treatment programme for childhood (≤16 years) low grade glioma - A final report. Eur J Cancer. 2017;81:206-225. doi:10.1016/j.ejca.2017.04.019. PMID:28649001; PMCID:PMC5517338.
Ater JL, Zhou T, Holmes E, Mazewski CM, Booth TN, Freyer DR, Lazarus KH, Packer RJ, Prados M, Sposto R, Vezina G, Wisoff JH, Pollack IF. Randomized study of two chemotherapy regimens for treatment of low-grade glioma in young children: a report from the Children’s Oncology Group. J Clin Oncol. 2012;30(21):2641-2647. doi:10.1200/jco.2011.36.6054. PMID:22665535; PMCID:PMC3413276.
Alvaro Lassaletta, Katrin Scheinemann, Shayna M. Zelcer, Juliette Hukin, Beverley A. Wilson, Nada Jabado, Anne Sophie Carret, Lucie Lafay-Cousin, et al. Phase II Weekly Vinblastine for Chemotherapy-Naïve Children With Progressive Low-Grade Glioma: A Canadian Pediatric Brain Tumor Consortium Study. JCO. 2016;34(29):3537-3543. doi:10.1200/jco.2016.68.1585.
Fangusaro J, Onar-Thomas A, Young Poussaint T, Wu S, Ligon AH, Lindeman N, Banerjee A, Packer RJ, Kilburn LB, Goldman S, Pollack IF, Qaddoumi I, Jakacki RI, Fisher PG, Dhall G, Baxter P, Kreissman SG, Stewart CF, Jones DTW, Pfister SM, Vezina G, Stern JS, Panigrahy A, Patay Z, Tamrazi B, Jones JY, Haque SS, Enterline DS, Cha S, Fisher MJ, Doyle LA, Smith M, Dunkel IJ, Fouladi M. Selumetinib in paediatric patients with BRAF-aberrant or neurofibromatosis type 1-associated recurrent, refractory, or progressive low-grade glioma: a multicentre, phase 2 trial. Lancet Oncol. 2019;20(7):1011-1022. doi:10.1016/s1470-2045(19)30277-3. PMID:31151904; PMCID:PMC6628202.
Anuradha Banerjee, Regina I. Jakacki, Arzu Onar-Thomas, Shengjie Wu, Theodore Nicolaides, Tina Young Poussaint, Jason Fangusaro, Joanna Phillips, et al. A phase I trial of the MEK inhibitor selumetinib (AZD6244) in pediatric patients with recurrent or refractory low-grade glioma: a Pediatric Brain Tumor Consortium (PBTC) study. Neuro-Oncology. 2017;19(8):1135-1144. doi:10.1093/neuonc/now282.
de Blank PMK, Orne-Ibekwe E, Packer R. International consensus recommendations for visual surveillance in optic pathway gliomas associated with neurofibromatosis type 1. J Neurooncol. 2020;148(3):571-578.
Listernick R, Louis DN, Packer RJ, Gutmann DH. Optic pathway gliomas in children with neurofibromatosis 1: consensus statement from the NF1 Optic Pathway Glioma Task Force. Ann Neurol. 1997;41(2):143-149. doi:10.1002/ana.410410204. PMID:9029062.
Makale metnini kopyalayıp tercih ettiğiniz yapay zeka asistanına yapıştırabilirsiniz.
Makale panoya kopyalandı
Aşağıdaki yapay zeka asistanlarından birini açın ve kopyalanan metni sohbet kutusuna yapıştırın.