O glioma do nervo óptico (optic nerve glioma / optic pathway glioma) é um tipo de glioma que se desenvolve no nervo óptico. Em sentido estrito, refere-se a gliomas que surgem no nervo óptico anterior ao quiasma óptico. Em sentido amplo, significa glioma que ocorre em toda a via óptica, incluindo a região posterior ao quiasma óptico (optic pathway glioma).
Histologicamente, a maioria é astrocitoma pilocítico benigno (pilocytic astrocytoma, WHO Grau I). No entanto, há relatos de casos malignos em uma minoria. Cerca de 70% dos casos ocorrem na infância, sendo uma doença rara que representa aproximadamente 0,5 a 5% dos tumores cerebrais pediátricos.
Há uma forte associação com a neurofibromatose tipo 1 (NF1, doença de von Recklinghausen), sendo que cerca de 20 a 30% dos casos de glioma do nervo óptico apresentam NF1 concomitante. Inversamente, o glioma do nervo óptico é a lesão orbitária mais comum em pacientes com NF1.
Classificação por localização e antecedente genético
Forma de ocorrência mais comum. Sintomas principais: perda visual unilateral e proptose.
Limitado ao nervo óptico dentro da órbita, a observação é a conduta básica.
Em casos associados a NF1, há relatos de regressão espontânea.
Tipo infiltrativo do quiasma óptico
Tipo que infiltra o quiasma óptico.
Causa deficiência visual binocular e o manejo torna-se complexo.
Ocorre frequentemente em idade precoce, sendo necessário avaliar a extensão para o hipotálamo.
Tipo via óptica-hipotalâmico
Tipo que se estende da região posterior ao quiasma óptico até o hipotálamo.
Pode estar associado a anormalidades endócrinas (distúrbios de crescimento, puberdade precoce, etc.).
O tratamento requer colaboração entre neurocirurgia e endocrinologia.
Quanto à classificação por background genético, existem o tipo associado à NF1 (cerca de 30% do total) e o tipo esporádico (cerca de 70%).
No tipo associado à NF1, também pode ocorrer envolvimento bilateral.
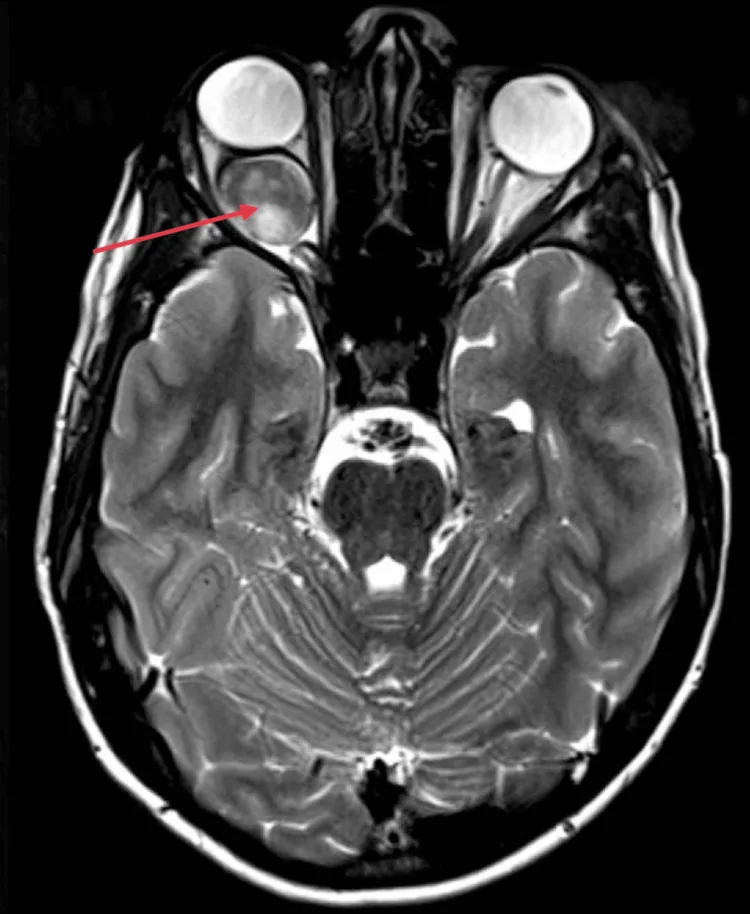
Kotecha MR, et al. Idiopathic Optic Nerve Glioma: A Case Report. Cureus. 2024. Figure 2. PMCID: PMC11744336. License: CC BY.
Na RM axial, o nervo óptico intraorbital direito apresenta aumento fusiforme, com realce intenso e homogêneo (seta vermelha), e observa-se proptose devido ao efeito de massa posterior ao globo ocular. Corresponde aos achados de imagem de RM (aumento homogêneo do nervo óptico e curvatura para baixo) discutidos na seção “2. Principais sintomas e achados clínicos”.
Crianças pequenas não relatam espontaneamente a diminuição da visão. Por isso, pais ou pessoas próximas frequentemente notam estrabismo (especialmente esotropia) e levam a criança ao oftalmologista pela primeira vez.
A diminuição da visão unilateral sem estrabismo é ainda mais facilmente negligenciada. Em alguns casos, já há atrofia do nervo óptico na primeira consulta.
O glioma óptico bilateral é mais comum em casos de início precoce. Geralmente é percebido por anormalidades no olhar ou comportamentos que indicam falta de visão, e o comprometimento visual pode ser grave.
A proptose não é evidente e não há dor.
QSe uma criança for diagnosticada com estrabismo, existe a possibilidade de ser glioma do nervo óptico?
A
No glioma do nervo óptico, crianças pequenas muitas vezes não conseguem perceber ou relatar diminuição da visão, então o estrabismo (especialmente esotropia) é frequentemente o primeiro sintoma que leva à consulta oftalmológica. Em crianças diagnosticadas com estrabismo, especialmente estrabismo unilateral, é importante considerar a possibilidade de glioma do nervo óptico e realizar exames complementares, incluindo acuidade visual, fundo de olho e exames de imagem.
O glioma óptico é a lesão orbitária mais comum na NF1
QPessoas com NF1 (neurofibromatose tipo 1) têm maior probabilidade de desenvolver glioma óptico?
A
Pacientes com NF1 apresentam risco significativamente maior de glioma óptico, e 20-30% de todos os casos de glioma óptico estão associados à NF11). Após o diagnóstico de NF1, recomenda-se acompanhamento oftalmológico regular como triagem para glioma óptico. Por outro lado, quando um glioma óptico é descoberto em uma criança, é importante verificar em todos os casos se os critérios diagnósticos para NF1 são atendidos.
Doença mais importante no diagnóstico diferencial.
Mais comum em mulheres adultas, podendo estar associada à NF2.
Na TC/RM, o sinal do trilho de bonde (tram-track sign) é característico e útil para diferenciar do glioma do nervo óptico.
Neurite óptica
Frequentemente de início agudo e associado a dor ao movimentar o olho.
A RM mostra realce de contraste do nervo óptico, mas o aumento de volume é leve.
Geralmente melhora com tratamento com corticosteroides.
O glioma do nervo óptico apresenta aumento uniforme e curvatura para baixo (downward kinking), que pode ser claramente distinguido do sinal do trilho de trem (tram-track sign) do meningioma da bainha do nervo óptico.
QQual é a diferença entre glioma do nervo óptico e meningioma da bainha do nervo óptico?
A
O glioma do nervo óptico é um tumor benigno (astrocitoma pilocítico) comum em crianças, podendo estar associado à NF1. Na TC/RM, é caracterizado por aumento uniforme do nervo óptico e curvatura para baixo (downward kinking). Já o meningioma da bainha do nervo óptico é mais frequente em mulheres adultas, podendo estar associado à NF2, e na TC/RM apresenta o sinal do trilho de trem (tram-track sign: calcificação ou realce ao redor do nervo óptico), o que permite a diferenciação.
Como o tumor é benigno e comum em crianças, quando limitado ao nervo óptico intraorbital, a ressecção cirúrgica e a radioterapia não são realizadas em princípio. A conduta básica é a observação cuidadosa com exames de imagem periódicos (RM a cada 3-6 meses).
No passado, a cirurgia era realizada, mas atualmente tende-se a evitar a ressecção cirúrgica devido ao alto risco de perda irreversível da visão. Em casos associados à NF1, há relatos de regressão espontânea, portanto a observação é especialmente cautelosa.
Quimioterapia (casos progressivos ou com perda visual)
Em casos de perda visual progressiva ou aumento tumoral, a quimioterapia combinada com carboplatina e vincristina (CV) é usada como tratamento padrão de primeira linha3)4).
Regime padrão de CV (COG A9952, etc.):
Vincristina: 1,5 mg/m² IV, 1x/semana por 10 semanas
Carboplatina: 550 mg/m² IV, a cada 3 semanas
A taxa de resposta objetiva (remissão parcial + estabilização) da terapia CV é relatada como 60-80%4).
Considerada em casos avançados resistentes à quimioterapia. No entanto, em crianças, há preocupações com risco de segundo câncer, disfunção endócrina (irradiação próxima ao hipotálamo) e impacto na função cognitiva, portanto, tende-se a evitar sempre que possível.
Os achados histopatológicos do glioma do nervo óptico são de astrocitoma pilocítico (pilocytic astrocytoma, WHO Grau I) benigno. É essencialmente diferente de gliomas de alto grau, como o glioblastoma multiforme (WHO Grau IV).
As células tumorais apresentam morfologia característica com processos celulares bipolares e contêm fibras de Rosenthal. Originam-se das células gliais (astrócitos) do nervo óptico, comprimindo e substituindo o nervo por dentro.
O glioma do nervo óptico associado à NF1 (neurofibromatose tipo 1) é causado por mutações no gene NF1 (cromossomo 17q11.2).
O gene NF1 é um gene supressor tumoral que codifica a proteína neurofibromina.
neurofibromina funciona como proteína ativadora de Ras-GTPase (GAP), suprimindo a sinalização de Ras
Mutação NF1 → perda de função da neurofibromina → ativação constitutiva da via de sinalização Ras → aumento da sinalização MAPK → proliferação descontrolada de células gliais
Em astrocitomas pilocíticos esporádicos (não associados a NF1), o gene de fusão BRAF-KIAA1549 é frequentemente encontrado. Esse gene de fusão também ativa a via MAPK, promovendo o crescimento tumoral.
Alguns casos apresentam a mutação BRAF V600E, e esses casos tendem a ter maior grau de malignidade6).
A maioria dos gliomas do nervo óptico é de baixo grau e apresenta crescimento lento.
O tumor aumenta o nervo óptico internamente, causando encurvamento (kinking/curvatura para baixo) dentro da órbita.
O aumento uniforme e a curvatura para baixo na RM são pontos-chave no diagnóstico por imagem.
O prognóstico visual não é uniforme, com casos progressivos e estáveis coexistindo, portanto, além do tamanho do tumor, é necessária uma avaliação longitudinal da função visual. Existem casos em que a função visual piora mesmo com achados de RM estáveis, e, inversamente, em casos associados a NF1, pode ocorrer regressão espontânea. 1, 8, 9)
Avaliação da função visual: medir repetidamente acuidade visual, campo visual, visão de cores e RAPD de acordo com a idade
Acompanhamento por imagem: rastrear recrescimento, extensão ao quiasma e extensão intracraniana por RM 8)
Avaliação endócrina: em casos de extensão hipotalâmica, verificar distúrbios de crescimento, puberdade precoce e diabetes insípido
Manejo sistêmico da NF1: realizar acompanhamento sistêmico incluindo lesões cutâneas, outros tumores e aspectos do desenvolvimento 2, 9)
A eficácia dos inibidores de MEK para gliomas de baixo grau associados à NF1 foi relatada.
No estudo SPRINT (Fase II), foi relatada uma taxa de resposta objetiva de 66% com selumetinibe para gliomas de baixo grau progressivos associados à NF1 (neurofibromas plexiformes)7).
A aplicação em gliomas de baixo grau associados à NF1, incluindo gliomas do nervo óptico, está sendo investigada.
Para gliomas de baixo grau pediátricos com mutação BRAF V600E, a terapia combinada de dabrafenibe + trametinibe está sendo avaliada em ensaios clínicos6).
Em casos com fusão BRAF-KIAA1549 positiva, a eficácia dos inibidores de BRAF é limitada.
Com o advento dos inibidores de MEK e BRAF, está ocorrendo uma mudança da quimioterapia convencional (terapia CV) para o tratamento personalizado baseado no perfil molecular8).
Futuramente, a seleção do tratamento baseada no perfil de mutações genéticas (fusão BRAF, BRAF V600E, mutação NF1, etc.) pode se tornar padronizada.
Listernick R, Charrow J, Greenwald M, Mets M. Natural history of optic pathway tumors in children with neurofibromatosis type 1: a longitudinal study. J Pediatr. 1994;125(1):63-66. doi:10.1016/s0022-3476(94)70122-9. PMID:8021787.
Ferner RE, Ferner RE, Huson SM, Thomas N, Moss C, Willshaw H, Evans DG, Upadhyaya M, Towers R, Gleeson M, Steiger C, Kirby A. Guidelines for the diagnosis and management of individuals with neurofibromatosis 1. J Med Genet. 2007;44(2):81-88. doi:10.1136/jmg.2006.045906. PMID:17105749; PMCID:PMC2598063.
Gnekow AK, Walker DA, Kandels D, Picton S, Giorgio Perilongo, Grill J, Stokland T, Sandstrom PE, Warmuth-Metz M, Pietsch T, Giangaspero F, Schmidt R, Faldum A, Kilmartin D, De Paoli A, De Salvo GL, of the Low Grade Glioma Consortium and the participating centers. A European randomised controlled trial of the addition of etoposide to standard vincristine and carboplatin induction as part of an 18-month treatment programme for childhood (≤16 years) low grade glioma - A final report. Eur J Cancer. 2017;81:206-225. doi:10.1016/j.ejca.2017.04.019. PMID:28649001; PMCID:PMC5517338.
Ater JL, Zhou T, Holmes E, Mazewski CM, Booth TN, Freyer DR, Lazarus KH, Packer RJ, Prados M, Sposto R, Vezina G, Wisoff JH, Pollack IF. Randomized study of two chemotherapy regimens for treatment of low-grade glioma in young children: a report from the Children’s Oncology Group. J Clin Oncol. 2012;30(21):2641-2647. doi:10.1200/jco.2011.36.6054. PMID:22665535; PMCID:PMC3413276.
Alvaro Lassaletta, Katrin Scheinemann, Shayna M. Zelcer, Juliette Hukin, Beverley A. Wilson, Nada Jabado, Anne Sophie Carret, Lucie Lafay-Cousin, et al. Phase II Weekly Vinblastine for Chemotherapy-Naïve Children With Progressive Low-Grade Glioma: A Canadian Pediatric Brain Tumor Consortium Study. JCO. 2016;34(29):3537-3543. doi:10.1200/jco.2016.68.1585.
Fangusaro J, Onar-Thomas A, Young Poussaint T, Wu S, Ligon AH, Lindeman N, Banerjee A, Packer RJ, Kilburn LB, Goldman S, Pollack IF, Qaddoumi I, Jakacki RI, Fisher PG, Dhall G, Baxter P, Kreissman SG, Stewart CF, Jones DTW, Pfister SM, Vezina G, Stern JS, Panigrahy A, Patay Z, Tamrazi B, Jones JY, Haque SS, Enterline DS, Cha S, Fisher MJ, Doyle LA, Smith M, Dunkel IJ, Fouladi M. Selumetinib in paediatric patients with BRAF-aberrant or neurofibromatosis type 1-associated recurrent, refractory, or progressive low-grade glioma: a multicentre, phase 2 trial. Lancet Oncol. 2019;20(7):1011-1022. doi:10.1016/s1470-2045(19)30277-3. PMID:31151904; PMCID:PMC6628202.
Anuradha Banerjee, Regina I. Jakacki, Arzu Onar-Thomas, Shengjie Wu, Theodore Nicolaides, Tina Young Poussaint, Jason Fangusaro, Joanna Phillips, et al. A phase I trial of the MEK inhibitor selumetinib (AZD6244) in pediatric patients with recurrent or refractory low-grade glioma: a Pediatric Brain Tumor Consortium (PBTC) study. Neuro-Oncology. 2017;19(8):1135-1144. doi:10.1093/neuonc/now282.
de Blank PMK, Orne-Ibekwe E, Packer R. International consensus recommendations for visual surveillance in optic pathway gliomas associated with neurofibromatosis type 1. J Neurooncol. 2020;148(3):571-578.
Listernick R, Louis DN, Packer RJ, Gutmann DH. Optic pathway gliomas in children with neurofibromatosis 1: consensus statement from the NF1 Optic Pathway Glioma Task Force. Ann Neurol. 1997;41(2):143-149. doi:10.1002/ana.410410204. PMID:9029062.
Copie o texto do artigo e cole no assistente de IA de sua preferência.
Artigo copiado para a área de transferência
Abra um assistente de IA abaixo e cole o texto copiado na conversa.