Ein Optikusgliom (Optic nerve glioma / Optic pathway glioma) ist eine Art von Gliom, das am Sehnerv auftritt. Im engeren Sinne bezeichnet es ein Gliom des Sehnervs vor dem Chiasma opticum. Im weiteren Sinne umfasst es ein Gliom des gesamten Sehwegs einschließlich des Bereichs hinter dem Chiasma opticum (Optic pathway glioma).
Histologisch handelt es sich meist um ein gutartiges pilozytisches Astrozytom (WHO Grad I). Es gibt jedoch auch Berichte über bösartige Fälle. Etwa 70% treten im Kindesalter auf, und es ist eine seltene Erkrankung, die etwa 0,5–5% der pädiatrischen Hirntumoren ausmacht.
Es besteht ein starker Zusammenhang mit der Neurofibromatose Typ 1 (NF1, Morbus Recklinghausen). Bei etwa 20–30% der Patienten mit Optikusgliom wird eine NF1 festgestellt. Umgekehrt ist das Optikusgliom die häufigste orbitale Läsion bei NF1-Patienten.
Klassifikation nach Lokalisation und genetischem Hintergrund
Häufigste Entstehungsform. Einseitige Sehverschlechterung und Exophthalmus sind die Hauptsymptome.
Begrenzt auf den Sehnerv innerhalb der Orbita, ist die Beobachtung die Grundlage. Bei NF1-assoziierten Fällen wurde auch eine spontane Regression berichtet.
Chiasma-invasiver Typ
Typ, der bis zum Chiasma opticum infiltriert.
Es kommt zu beidseitigen Sehstörungen, und die Behandlung wird komplex.
Es tritt häufig in jungen Jahren auf, und die Ausdehnung in den Hypothalamus muss beurteilt werden.
Sehbahn-Hypothalamus-Typ
Typ, der sich vom hinteren Chiasma bis zum Hypothalamus ausbreitet.
Es können endokrine Störungen (Wachstumsstörungen, vorzeitige Pubertät usw.) auftreten.
Die Behandlung erfordert eine Zusammenarbeit zwischen Neurochirurgie und Endokrinologie.
Nach genetischem Hintergrund gibt es den NF1-assoziierten Typ (ca. 30% aller Fälle) und den sporadischen Typ (ca. 70%).
Beim NF1-assoziierten Typ kann auch eine bilaterale Manifestation auftreten.
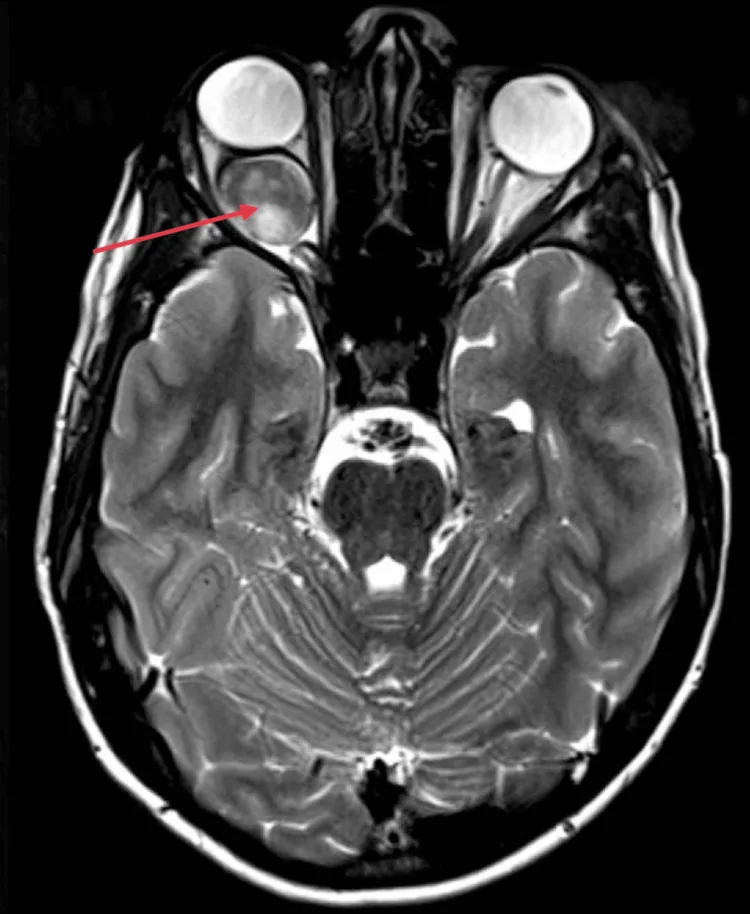
Kotecha MR, et al. Idiopathic Optic Nerve Glioma: A Case Report. Cureus. 2024. Figure 2. PMCID: PMC11744336. License: CC BY.
Axiales MRT zeigt eine spindelförmige Verdickung des rechten intraorbitalen Sehnervs mit homogener, starker Kontrastmittelanreicherung (roter Pfeil) und Exophthalmus durch raumfordernde Wirkung hinter dem Bulbus. Dies entspricht den MRT-Befunden (gleichmäßige Verdickung des Sehnervs, downward kinking), die im Abschnitt „2. Hauptsymptome und klinische Befunde“ behandelt werden.
Kleinkinder klagen nicht selbst über Sehverschlechterung. Daher bemerken Eltern oder Bezugspersonen häufig Schielen (insbesondere Einwärtsschielen) und suchen erstmals einen Augenarzt auf.
Einseitige Sehverschlechterung ohne Schielen wird noch später entdeckt. Bei der Erstuntersuchung kann bereits eine Optikusatrophie vorliegen.
Beidseitige Optikusgliome treten häufiger bei jungen Kindern auf. Sie werden erst durch Auffälligkeiten der Blickrichtung oder „Nicht-Sehen-Verhalten“ bemerkt, und die Sehbehinderung kann schwerwiegend sein.
Ein Exophthalmus ist nicht offensichtlich, und es treten keine Schmerzen auf.
QKann ein Sehnervengliom vorliegen, wenn bei einem Kind Schielen diagnostiziert wird?
A
Bei einem Sehnervengliom können kleine Kinder eine Sehverschlechterung oft nicht selbst bemerken oder äußern, sodass Schielen (insbesondere Einwärtsschielen) als erstes Symptom häufig der Grund für einen Augenarztbesuch ist. Bei Kindern mit diagnostiziertem Schielen, insbesondere bei einseitigem Schielen, ist es wichtig, ein Sehnervengliom in Betracht zu ziehen und eine gründliche Untersuchung einschließlich Sehtest, Augenhintergrunduntersuchung und Bildgebung zu erwägen.
Bei orbitalen Läsionen der NF1 ist das Optikusgliom am häufigsten
QErhöht NF1 (Neurofibromatose Typ 1) das Risiko für ein Optikusgliom?
A
Patienten mit NF1 haben ein deutlich erhöhtes Risiko für ein Optikusgliom; 20–30 % aller Optikusgliomfälle treten in Verbindung mit NF1 auf 1). Nach der NF1-Diagnose wird eine regelmäßige augenärztliche Nachsorge als Screening auf Optikusgliome empfohlen. Umgekehrt ist es wichtig, bei jedem Kind mit einem neu entdeckten Optikusgliom zu prüfen, ob die Diagnosekriterien für NF1 erfüllt sind.
Eine Biopsie ist in der Regel nicht erforderlich (Diagnose mittels Bildgebung möglich). Die Gewebebestätigung erfolgt bei der chirurgischen Entfernung.
Tritt häufiger bei erwachsenen Frauen auf, assoziiert mit NF2.
CT/MRT zeigt charakteristisches Tram-Track-Zeichen, hilfreich zur Abgrenzung vom Optikusgliom.
Optikusneuritis
Häufig akuter Beginn und Schmerzen bei Augenbewegungen.
Im MRT zeigt sich eine Kontrastmittelanreicherung des Sehnervs, jedoch nur eine geringe Schwellung.
Die Symptome bessern sich meist unter Steroidtherapie.
Das Optikusgliom zeigt eine gleichmäßige Verdickung und Downward-Kinking, die sich klar vom Tram-Track-Zeichen des Optikusscheidenmeningioms unterscheiden lässt.
QWas ist der Unterschied zwischen einem Optikusgliom und einem Optikusscheidenmeningiom?
A
Das Optikusgliom ist ein gutartiger Tumor (pilozytisches Astrozytom), der häufig bei Kindern auftritt und mit NF1 assoziiert sein kann. Im CT/MRT zeigt es eine gleichmäßige Verdickung des Sehnervs und ein Downward-Kinking. Das Optikusscheidenmeningiom tritt häufiger bei erwachsenen Frauen auf, kann mit NF2 assoziiert sein und zeigt im CT/MRT ein Tram-Track-Zeichen (verkalkte oder kontrastmittelanreichernde Linien entlang des Sehnervs), was zur Unterscheidung dient.
Da der Tumor gutartig ist und häufig bei Kindern auftritt, wird bei einer auf den Sehnerv in der Augenhöhle beschränkten Lage grundsätzlich keine chirurgische Resektion oder Strahlentherapie durchgeführt. Die Grundstrategie ist eine sorgfältige Beobachtung mit regelmäßigen bildgebenden Untersuchungen (MRT alle 3–6 Monate).
Früher wurde operiert, aber aufgrund des hohen Risikos eines irreversiblen Sehverlusts wird heute eher auf eine chirurgische Resektion verzichtet. Bei NF1-assoziierten Fällen gibt es Berichte über spontane Regression, weshalb eine besonders sorgfältige Beobachtung erfolgt.
Chemotherapie (bei Fortschreiten oder Sehverschlechterung)
Bei fortschreitender Sehverschlechterung oder Tumorwachstum wird eine Kombinationschemotherapie mit Carboplatin und Vincristin (CV-Therapie) als Standard-Erstlinientherapie eingesetzt3)4).
Standard-CV-Regime (COG A9952 etc.):
Vincristin: 1,5 mg/m² i.v., einmal wöchentlich × 10 Wochen
Carboplatin: 550 mg/m² i.v., alle 3 Wochen
Die objektive Ansprechrate (partielle Remission + Stabilisierung) der CV-Therapie wird mit 60–80 % angegeben4).
Optionen der Zweitlinientherapie:
Cisplatin + Etoposid
Temozolomid (Alkylanzien)
Vinblastin-Monotherapie (bei Rezidiv nach CV-Therapie5))
Wird bei chemotherapieresistenten fortgeschrittenen Fällen in Betracht gezogen. Bei Kindern wird sie jedoch aufgrund des Risikos von Zweitmalignomen, endokrinen Störungen (bei Bestrahlung in der Nähe des Hypothalamus) und Auswirkungen auf die kognitive Funktion nach Möglichkeit vermieden.
Die histopathologischen Befunde des Sehnervenglioms sind ein benignes pilozytisches Astrozytom (pilocytic astrocytoma, WHO Grad I). Es unterscheidet sich grundlegend von hochgradigen Gliomen wie dem Glioblastoma multiforme (WHO Grad IV).
Die Tumorzellen zeigen eine charakteristische Morphologie mit bipolaren Zellfortsätzen und enthalten Rosenthal-Fasern. Sie entstehen aus Gliazellen (Astrozyten) des Sehnervs und komprimieren und ersetzen den Sehnerv von innen.
Bei sporadischen (nicht mit NF1 assoziierten) pilozytischen Astrozytomen tritt häufig das BRAF-KIAA1549-Fusionsgen auf. Dieses Fusionsgen aktiviert ebenfalls den MAPK-Signalweg und fördert das Tumorwachstum.
Einige Fälle weisen die BRAF V600E-Mutation auf, die tendenziell mit einem höheren Malignitätsgrad einhergeht6).
Die Mehrheit der Sehnervengliome sind niedriggradig (low-grade) und zeigen ein langsames Wachstum. Der Tumor schwillt den Sehnerv von innen an und verursacht eine Abknickung (Kinking, Downward-Kinking) des Nervs in der Orbita. Die gleichmäßige Schwellung und Abwärtsknickung im MRT sind Schlüsselpunkte der Bilddiagnostik.
Die Sehprognose ist uneinheitlich, da fortschreitende und stabile Fälle nebeneinander existieren. Daher ist nicht nur die Tumorgröße, sondern auch eine Längsschnittbewertung der Sehfunktion erforderlich. Es gibt Fälle, in denen sich die Sehfunktion trotz stabiler MRT-Befunde verschlechtert, während bei NF1-assoziierten Fällen eine spontane Regression auftreten kann. 1, 8, 9)
Sehfunktionsbeurteilung: Sehschärfe, Gesichtsfeld, Farbsehen und RAPD altersgerecht wiederholt messen
Bildgebende Verlaufskontrolle: Mittels MRT Nachwachsen, Chiasmaausdehnung und intrakranielle Ausdehnung verfolgen8)
Endokrine Beurteilung: Bei hypothalamischer Ausdehnung Wachstumsstörungen, vorzeitige Pubertät und Diabetes insipidus einschließen
NF1-Ganzkörpermanagement: Hautveränderungen, andere Tumore und Entwicklungsaspekte parallel verfolgen2, 9)
Die Wirksamkeit von MEK-Inhibitoren bei NF1-assoziierten niedriggradigen Gliomen wurde berichtet.
In der SPRINT-Studie (Phase II) wurde eine objektive Ansprechrate von 66 % für Selumetinib bei NF1-assoziierten progressiven niedriggradigen Gliomen (plexiforme Neurofibrome) berichtet 7).
Die Anwendung bei NF1-assoziierten niedriggradigen Gliomen, einschließlich Optikusgliomen, wird untersucht.
Bei pädiatrischen niedriggradigen Gliomen mit BRAF V600E-Mutation wird die Kombinationstherapie mit Dabrafenib + Trametinib in klinischen Studien evaluiert6).
Bei BRAF-KIAA1549-Fusions-positiven Fällen ist die Wirksamkeit von BRAF-Inhibitoren begrenzt.
Mit dem Aufkommen von MEK-Inhibitoren und BRAF-Inhibitoren findet ein Wandel von der konventionellen Chemotherapie (CV-Therapie) hin zu einer individualisierten Behandlung basierend auf dem molekularen Profil statt8).
Zukünftig könnte die Therapieauswahl basierend auf dem genetischen Mutationsprofil (BRAF-Fusion, BRAF V600E, NF1-Mutation usw.) standardisiert werden.
Listernick R, Charrow J, Greenwald M, Mets M. Natural history of optic pathway tumors in children with neurofibromatosis type 1: a longitudinal study. J Pediatr. 1994;125(1):63-66. doi:10.1016/s0022-3476(94)70122-9. PMID:8021787.
Ferner RE, Ferner RE, Huson SM, Thomas N, Moss C, Willshaw H, Evans DG, Upadhyaya M, Towers R, Gleeson M, Steiger C, Kirby A. Guidelines for the diagnosis and management of individuals with neurofibromatosis 1. J Med Genet. 2007;44(2):81-88. doi:10.1136/jmg.2006.045906. PMID:17105749; PMCID:PMC2598063.
Gnekow AK, Walker DA, Kandels D, Picton S, Giorgio Perilongo, Grill J, Stokland T, Sandstrom PE, Warmuth-Metz M, Pietsch T, Giangaspero F, Schmidt R, Faldum A, Kilmartin D, De Paoli A, De Salvo GL, of the Low Grade Glioma Consortium and the participating centers. A European randomised controlled trial of the addition of etoposide to standard vincristine and carboplatin induction as part of an 18-month treatment programme for childhood (≤16 years) low grade glioma - A final report. Eur J Cancer. 2017;81:206-225. doi:10.1016/j.ejca.2017.04.019. PMID:28649001; PMCID:PMC5517338.
Ater JL, Zhou T, Holmes E, Mazewski CM, Booth TN, Freyer DR, Lazarus KH, Packer RJ, Prados M, Sposto R, Vezina G, Wisoff JH, Pollack IF. Randomized study of two chemotherapy regimens for treatment of low-grade glioma in young children: a report from the Children’s Oncology Group. J Clin Oncol. 2012;30(21):2641-2647. doi:10.1200/jco.2011.36.6054. PMID:22665535; PMCID:PMC3413276.
Alvaro Lassaletta, Katrin Scheinemann, Shayna M. Zelcer, Juliette Hukin, Beverley A. Wilson, Nada Jabado, Anne Sophie Carret, Lucie Lafay-Cousin, et al. Phase II Weekly Vinblastine for Chemotherapy-Naïve Children With Progressive Low-Grade Glioma: A Canadian Pediatric Brain Tumor Consortium Study. JCO. 2016;34(29):3537-3543. doi:10.1200/jco.2016.68.1585.
Fangusaro J, Onar-Thomas A, Young Poussaint T, Wu S, Ligon AH, Lindeman N, Banerjee A, Packer RJ, Kilburn LB, Goldman S, Pollack IF, Qaddoumi I, Jakacki RI, Fisher PG, Dhall G, Baxter P, Kreissman SG, Stewart CF, Jones DTW, Pfister SM, Vezina G, Stern JS, Panigrahy A, Patay Z, Tamrazi B, Jones JY, Haque SS, Enterline DS, Cha S, Fisher MJ, Doyle LA, Smith M, Dunkel IJ, Fouladi M. Selumetinib in paediatric patients with BRAF-aberrant or neurofibromatosis type 1-associated recurrent, refractory, or progressive low-grade glioma: a multicentre, phase 2 trial. Lancet Oncol. 2019;20(7):1011-1022. doi:10.1016/s1470-2045(19)30277-3. PMID:31151904; PMCID:PMC6628202.
Anuradha Banerjee, Regina I. Jakacki, Arzu Onar-Thomas, Shengjie Wu, Theodore Nicolaides, Tina Young Poussaint, Jason Fangusaro, Joanna Phillips, et al. A phase I trial of the MEK inhibitor selumetinib (AZD6244) in pediatric patients with recurrent or refractory low-grade glioma: a Pediatric Brain Tumor Consortium (PBTC) study. Neuro-Oncology. 2017;19(8):1135-1144. doi:10.1093/neuonc/now282.
de Blank PMK, Orne-Ibekwe E, Packer R. International consensus recommendations for visual surveillance in optic pathway gliomas associated with neurofibromatosis type 1. J Neurooncol. 2020;148(3):571-578.
Listernick R, Louis DN, Packer RJ, Gutmann DH. Optic pathway gliomas in children with neurofibromatosis 1: consensus statement from the NF1 Optic Pathway Glioma Task Force. Ann Neurol. 1997;41(2):143-149. doi:10.1002/ana.410410204. PMID:9029062.
Kopieren Sie den Artikeltext und fügen Sie ihn in den KI-Assistenten Ihrer Wahl ein.
Artikel in die Zwischenablage kopiert
Öffnen Sie unten einen KI-Assistenten und fügen Sie den kopierten Text in den Chat ein.