Das Rhabdomyosarkom ist ein bösartiger Tumor, der von mesenchymalen Zellen der Orbita ausgeht und eine Differenzierung in quergestreifte Muskulatur zeigt. Es entsteht aus undifferenzierten mesenchymalen Zellen der Orbita, nicht aus der malignen Transformation von differenziertem Muskelgewebe wie den äußeren Augenmuskeln. Es ist der häufigste bösartige Orbitaltumor bei Kindern; bei einem schnell wachsenden Orbitaltumor bei einem Kind sollte man zuerst an diese Erkrankung denken.
In den USA macht es etwa 5 % aller pädiatrischen Krebserkrankungen aus, mit etwa 250 Neudiagnosen pro Jahr. Davon sind etwa 10 % (etwa 35 Fälle pro Jahr) orbitalen Ursprungs. Die jährliche Inzidenz wird auf 4 Fälle pro Million Einwohner in der Gesamtbevölkerung und 4,5 Fälle pro Million Kinder geschätzt1).
Das Durchschnittsalter bei Diagnose beträgt 7–8 Jahre; zwei Drittel der Fälle treten vor dem 10. Lebensjahr auf. 90 % der Fälle treten vor dem 16. Lebensjahr auf, mit einer leichten Bevorzugung von Jungen (Geschlechterverhältnis 5:3). Es gibt keine Unterschiede in der Inzidenz nach Rasse. Der Tumor ist gut abgegrenzt und tritt häufig im oberen Teil der Orbita auf.
Die Verteilung der Lokalisationen des okulären Rhabdomyosarkoms ist: Orbita 76 %, Konjunktiva 12 %, Uvea 9 %, Augenlid 3 %. Es kann innerhalb der Orbita entstehen oder von umgebendem Gewebe wie den Nasennebenhöhlen ausgehen und in die Orbita einwachsen. Die Orbita ist der primäre Ursprungsort von 10 % aller Rhabdomyosarkome; andere Lokalisationen umfassen den Urogenitaltrakt (22 %), die Extremitäten (18 %), parameningeale Regionen (16 %) und andere Kopf-Hals-Bereiche (10 %).
QIn welchem Alter tritt das Rhabdomyosarkom am häufigsten auf?
A
Zwei Drittel der Fälle treten bei Kindern unter 10 Jahren auf, das Durchschnittsalter liegt bei 7–8 Jahren. 90 % der Fälle treten vor dem 16. Lebensjahr auf. Ein Auftreten bei Erwachsenen ist selten, wurde aber berichtet.
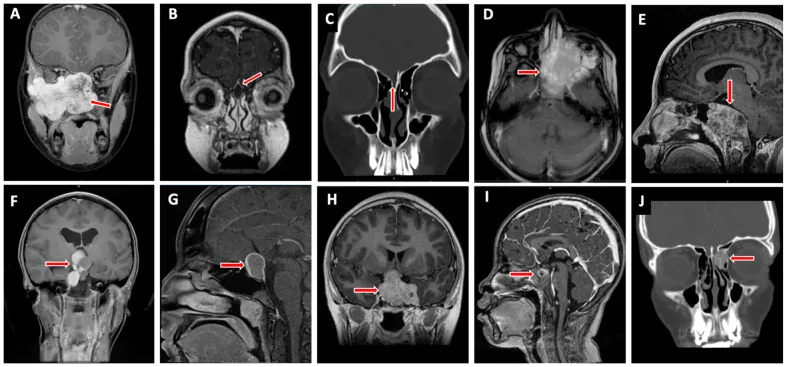
Valencia-Sanchez BA, et al. Special Considerations in Pediatric Endoscopic Skull Base Surgery. J Clin Med. 2024. Figure 2. PMCID: PMC11013018. License: CC BY.
Der rote Pfeil zeigt ein Rhabdomyosarkom (D), eine der typischen Läsionen der Schädelbasis bei Kindern. Dies entspricht der Tumorinfiltration, die im Abschnitt „2. Hauptsymptome und klinische Befunde“ behandelt wird.
Schneller Exophthalmus: Ein innerhalb weniger Wochen schnell fortschreitender einseitiger Exophthalmus ist ein typisches Symptom.
Lidschwellung: Mit dem Wachstum des Tumors schwillt das Augenlid an. Selbst bei starker Schwellung ist die Rötung gering, Entzündungszeichen sind spärlich und die Schmerzen sind nicht stark, was charakteristisch ist.
Einschränkung der Augenbeweglichkeit, Ptosis usw., die je nach Lokalisation des Tumors unterschiedliche klinische Bilder zeigen.
Tritt häufig bei Kindern im Alter von 0–9 Jahren auf und ist durch eine schnelle Progression gekennzeichnet.
Die Läsionen treten häufig oben und oben-innen auf, wobei der Tumor den Augapfel nach unten und außen verlagert. Außerhalb des Muskelkegels findet sich ein relativ scharf begrenzter, runder bis ovaler Tumor.
Etwa 10 % der Fälle gehen von den Nasennebenhöhlen und der Nasenhöhle aus und dringen sekundär in die Orbita ein; sie können sich mit Sinusitis, Nasenverstopfung oder Nasenbluten äußern. Hintere Tumoren können mit Aderhautfalten, Netzhautablösung oder Papillenödem einhergehen.
Bei primärer Bindehautbeteiligung erscheint der Tumor als granulomatöse, rosafarbene, traubenartige Masse im Bindehautfornix. Bei primärem Uveabefall zeigt er sich als Irismasse und kann mit Vorderkammeraussaat oder sekundärem Glaukom einhergehen.
Metastasen treten am häufigsten in der Lunge auf, gefolgt von Knochenmark, Knochen und Lymphknoten. Die Orbita enthält fast keine Lymphgefäße, aber vordere Tumoren der Bindehaut und der Lider können in regionäre Lymphknoten metastasieren.
QWelche Erkrankungen können mit einem Rhabdomyosarkom verwechselt werden?
A
Zu den klinischen Differentialdiagnosen gehören Neuroblastom, Chlorom, Lymphangiom, infantiles Hämangiom, Orbitalphlegmone und unspezifische Entzündung. Darüber hinaus umfassen die Differentialdiagnosen auch die orbitale Ausdehnung einer Sinusitis, Ruptur einer Dermoidzyste, Ewing-Sarkom und leukämische Orbitainfiltration. Die Differenzierung erfolgt anhand des raschen Fortschreitens, der Bildgebung und der Biopsiebefunde.
Das Rhabdomyosarkom entsteht nicht aus den äußeren Augenmuskeln, sondern aus pluripotenten undifferenzierten mesenchymalen Zellen im orbitalen Weichgewebe. Die meisten Fälle sind sporadisch, und die genaue Ursache ist unbekannt.
Die damit verbundenen genetischen Risikofaktoren sind wie folgt:
Eine notfallmäßige CT des Kopfes und der Orbita wird durchgeführt, um das Vorhandensein eines Tumors in der Orbita, seine Homogenität, eine Zerstörung der Orbitawand sowie eine intrakranielle oder sinusale Ausdehnung zu überprüfen. Anschließend wird eine MRT durchgeführt, um die innere Beschaffenheit des Tumors und seine Lagebeziehung zum Augapfel und zu den äußeren Augenmuskeln zu beurteilen.
Die charakteristischen Befunde der einzelnen Modalitäten sind wie folgt:
Untersuchung
Hauptbefunde
CT
Homogener, scharf begrenzter runder bis ovaler Tumor. Moderate bis starke Kontrastmittelaufnahme.
MRT T1
Hypointens zum orbitalen Fett, isointens zu den äußeren Augenmuskeln.
MRT T2
Hyperintens zum orbitalen Fett und zu den äußeren Augenmuskeln.
Im MRT kann die Abgrenzung zum kapillären Hämangiom schwierig sein, aber das kapilläre Hämangiom ist reich an Blutgefäßen (Blutfluss), was eine Unterscheidung ermöglicht. Bei reichlich interstitieller Komponente unterscheidet sich die MRT-Signalintensität.
Zur Metastasensuche gehören Röntgen-Thorax, Knochenszintigraphie und Zytologie aus Knochenmarkaspiration. Für die Ganzkörpersuche werden auch PET/CT, Ganzkörper-CT und Szintigraphie eingesetzt.
Zur definitiven Diagnose ist eine Biopsie erforderlich. Eine Feinnadelaspirationsbiopsie ist unzureichend; es wird eine Exzisionsbiopsie oder Inzisionsbiopsie durchgeführt.
Exzisionsbiopsie: Wird gewählt, wenn eine chirurgische Entfernung ohne Schädigung wichtiger Strukturen möglich ist.
Inzisionsbiopsie: Wird gewählt, wenn der Tumor groß und in der hinteren Orbita lokalisiert ist.
Die intraoperative Schnellschnittdiagnose kann die Unterscheidung von anderen Tumoren erschweren. Grundsätzlich wird eine Biopsie im Notfall durchgeführt und frühzeitig mit Chemotherapie und Strahlentherapie begonnen.
Der Nachweis von Rhabdomyoblasten mittels Lichtmikroskopie, Immunhistochemie und Elektronenmikroskopie steht im Zentrum der Diagnose.
Querstreifung wird mittels HE-Färbung oder Masson-Trichrom-Färbung nachgewiesen. Zur Differenzierung undifferenzierter Zellen wird eine immunhistochemische Färbung durchgeführt; ein Muster von Desmin-positiv, HHF-35 (Actin)-positiv und α-glattmuskuläres Actin-negativ ist für die Diagnose nützlich. Elektronenmikroskopisch werden Aktin-Myosin-Bündel sowie A-, I- und Z-Band-Proteine nachgewiesen.
Klinische Differenzialdiagnosen: Neuroblastom, Chlorom, Lymphangiom, infantiles Hämangiom, Orbitazellulitis, unspezifische Entzündung. Darüber hinaus sind auch orbitale Ausdehnung einer Sinusitis, Orbitaphlegmone, Ruptur einer Dermoidzyste, intratumorale Blutung eines Hämangioms, Blutung eines Lymphangioms, Ewing-Sarkom, orbitale Infiltration bei Leukämie (Chlorom) und Orbitablutung zu berücksichtigen.
Eine vollständige chirurgische Entfernung ist nicht möglich und sollte nicht angestrebt werden. Derzeit besteht das Hauptziel der Chirurgie in der Sicherung der Gewebediagnose; die Ära der Orbitaeviszeration als erste Wahl ist vorbei. Nach Bestätigung der pathologischen Diagnose durch eine Exzisionsbiopsie werden in der Pädiatrie ein Thorax-Abdominal-CT und eine Knochenmarkbiopsie durchgeführt, um das Vorliegen von Fernmetastasen, z. B. in der Lunge, zu überprüfen. Die systemische Chemotherapie ist die Grundlage der Behandlung; eine Dreifachtherapie aus Operation, Strahlentherapie und Chemotherapie ist die Standardbehandlung.
Bei der Chemotherapie wird hauptsächlich die VAC-Therapie (Vincristin + Actinomycin D + Cyclophosphamid) eingesetzt. Auch Ifosfamid und Etoposid haben sich als vorteilhaft erwiesen.
Die Strahlentherapie wird mit etwa 40–60 Gy durchgeführt. Seit April 2016 ist die Protonentherapie von der Krankenkasse abgedeckt und hat sich als Standardbehandlungsoption etabliert. Die Protonentherapie hat den Vorteil, die Dosis für das normale Gewebe zu reduzieren. In einigen Fällen wird auch eine hämatopoetische Stammzelltransplantation durchgeführt.
Das orbitale Rhabdomyosarkom wird nach der Stadieneinteilung der Intergroup Rhabdomyosarcoma Study (IRS) behandelt. In einer Serie von 30 Fällen von Shields et al. betrug die Verteilung: Gruppe I 7 %, Gruppe II 37 %, Gruppe III 53 %, Gruppe IV 3 %.
Gruppe
Definition
Behandlungsstrategie
I
Vollständige Resektion der lokalen Erkrankung
Nur Chemotherapie
II
Resttumor oder regionäre Lymphknotenmetastasen
Chemotherapie + Strahlentherapie
III
Inkomplette Resektion oder makroskopischer Resttumor
Chemotherapie + Strahlentherapie
IV
Fernmetastasen
Chemotherapie + Strahlentherapie
Für die Gruppen II bis IV wird die Strahlentherapie mit 4000–5000 cGy (40–50 Gy) über 4–5 Wochen verabreicht.
Bei nicht vollständig resezierbaren Fällen oder Rezidiven wird eine palliative Strahlentherapie oder Chemotherapie in Betracht gezogen. Chirurgisch kann eine Tumorentfernung oder Orbitaeviszeration erfolgen. Auch bei vollständiger Resektion ist eine Chemotherapie obligatorisch. Die Orbita ist eine prognostisch günstige Lokalisation; bei angemessener Behandlung wird ein Überleben von über 90 % erwartet.
Bei Infiltration in Schädel oder Nasennebenhöhlen oder Metastasen in Lunge, anderen Organen oder Halslymphknoten ist die Prognose schlecht. Bei frühzeitiger Erkennung und frühzeitigem Beginn der Chemotherapie ist die Prognose günstig.
Zu den ophthalmologischen Komplikationen der Chemotherapie gehören die durch Cyclophosphamid verursachte Keratokonjunktivitis sicca und Blepharokonjunktivitis, die durch Ifosfamid verursachte Konjunktivitis und verschwommenes Sehen sowie der durch Etoposid verursachte Zentralarterienverschluss der Netzhaut.
Lokalrezidive werden durch regelmäßige MRT-Untersuchungen beurteilt. Häufig treten hämatogene Metastasen auf, daher sind regelmäßige systemische Untersuchungen erforderlich.
Die langfristigen Sehergebnisse nach Behandlung (Shields) sind: 20/20 bis 20/40 bei 39 %, 20/50 bis 20/100 bei 18 % und 20/200 bis keine Lichtwahrnehmung bei 43 %.
QWelche Augenkomplikationen können nach der Behandlung eines Rhabdomyosarkoms auftreten?
Das Rhabdomyosarkom wird nach der WHO-Klassifikation in vier Subtypen eingeteilt: embryonal, alveolar, pleomorph und spindelzellig/sklerosierend 2).
Embryonaler Typ
Häufigkeit: 50–60 % aller Rhabdomyosarkome, 80–84 % der orbitalen Rhabdomyosarkome.
Histologie: Bündel spindelförmiger Zellen (fusiforme Zellen) in verschiedenen Richtungen. Kleine runde oder spindelförmige Zellen mit geringem nukleärem Pleomorphismus. Zytoplasma spärlich mit eosinophilen Granula, gestreifte Rhabdomyoblasten erkennbar. Häufiger im unteren Orbitabereich.
Prognose: Die 5-Jahres-Überlebensrate für orbitale Fälle beträgt 95 %, günstig. Durchschnittliches Erkrankungsalter 7–8 Jahre.
Alveolärer Typ
Häufigkeit: Etwa 20 % aller Rhabdomyosarkome. Etwa 10 % der orbitalen Rhabdomyosarkome.
Histologie: Tumor wächst in alveolären Mustern. Große unregelmäßige Zellen mit großem Kern und reichlich eosinophilem Zytoplasma, angeordnet um fibröse Trabekel. Neigung zu ausgedehnten Metastasen.
Prognose: Die 5-Jahres-Überlebensrate für orbitale Fälle beträgt 74 %. Dies ist der bösartigste und prognostisch ungünstigste histologische Typ.
Pleomorpher (differenzierter) Typ
Häufigkeit: Selten bei Kindern. Hauptsächlich an den Extremitäten Erwachsener (besonders Oberschenkel).
Histologie: Mehrkernige, runde bis längliche Zellen mit reichlich Zytoplasma, in denen Streifen erkennbar sind. Besteht aus hochgradig pleomorphen Tumorzellen. Selten, aber hoch differenziert.
Prognose: Bei orbitalen Fällen relativ günstig.
Der botryoide Typ ist eine Variante des embryonalen Typs, die häufig bei Säuglingen vorkommt und durch subepitheliale Tumorzellaggregate gekennzeichnet ist, die ein „traubenartiges“ Aussehen verleihen.
Histologisch haben der alveoläre und der pleomorphe Typ im Vergleich zum embryonalen Typ eine schlechtere Prognose. Die 5-Jahres-Überlebensrate für das gesamte Rhabdomyosarkom beträgt 66 %, aber das orbitale Rhabdomyosarkom wird aufgrund seiner Lokalisation in die Gruppe mit günstiger Prognose eingestuft.
Das alveoläre Rhabdomyosarkom ist durch wiederkehrende Chromosomentranslokationen t(2;13)(q35;q14) und t(1;13)(p36;q14) gekennzeichnet. Etwa 80 % der alveolären Rhabdomyosarkome weisen PAX3-FOXO1- oder PAX7-FOXO1-Translokationen auf. Die PAX3-FOXO1-Fusion ist mit hoher OLIG2-Expression und schlechter Prognose assoziiert2).
Das embryonale Rhabdomyosarkom weist keine wiederkehrenden strukturellen Chromosomenumlagerungen auf, aber ein häufiger Allelverlust auf Chromosom 11 (insbesondere der Region 11p15.5) wird beobachtet.
QWelche Unterschiede gibt es in der Prognose zwischen dem embryonalen und dem alveolären Typ?
A
Beim orbitalen Rhabdomyosarkom beträgt die 5-Jahres-Überlebensrate für den embryonalen Typ 95 %, während sie für den alveolären Typ mit 74 % niedriger ist. Der alveoläre Typ ist mit Chromosomentranslokationen wie PAX3-FOXO1 assoziiert und ein histologischer Typ, der zu ausgedehnten Metastasen neigt.
7. Aktuelle Forschung und zukünftige Perspektiven (Berichte aus der Forschungsphase)
In letzter Zeit wurde ein neuer Subtyp namens epitheloides Spindelzell-Rhabdomyosarkom mit TFCP2-Umlagerung identifiziert.
Li et al. (2023) berichteten, dass das epitheloide Spindelzell-Rhabdomyosarkom bevorzugt in Knochen (Kopf-Hals-Bereich, Becken) auftritt und durch EWSR1-TFCP2- oder FUS-TFCP2-Fusionen gekennzeichnet ist, ein Subtyp mit extrem schlechter Prognose3). Die berichtete mediane Überlebenszeit beträgt nur 17 Monate.
Molekulare zielgerichtete Therapie gegen PAX3-FOXO1
Die Forschung zu Therapien, die auf das PAX3-FOXO1-Fusionsprotein abzielen, das an etwa 80 % der alveolären Rhabdomyosarkome beteiligt ist, schreitet voran.
Ein Präparat, das kleine interferierende RNAs (siRNA) in Liposom-Protamin-Partikeln enthält, wurde berichtet, um die Expression von PAX3-FOXO1 in alveolären Rhabdomyosarkom-Zelllinien in vitro effizient herunterzuregulieren und zu einer Wachstumsverzögerung und -unterdrückung von alveolären Rhabdomyosarkom-Xenotransplantattumoren zu führen1).
Die Anwendung von Immun-Checkpoint-Inhibitoren (wie Nivolumab) bei metastasierten Sarkomen wird versucht1). Derzeit ist ihre Wirksamkeit gegen Rhabdomyosarkom jedoch nicht etabliert.
Behandlung des primären Rhabdomyosarkoms bei Erwachsenen
Eine Literaturübersicht über das primäre alveoläre Rhabdomyosarkom der Pinealisregion bei Erwachsenen deutet auf einen Zusammenhang zwischen Behandlungsintensität und Überleben hin.
In einer Übersicht von Chang et al. (2025) über 13 Fälle von primärem alveolärem Rhabdomyosarkom der Pinealisregion bei Erwachsenen betrug die mittlere Überlebenszeit mit alleiniger Operation etwa 5 Monate, mit Operation plus Strahlentherapie etwa 10,28 Monate und mit Operation plus Strahlentherapie plus Chemotherapie etwa 11,33 Monate, was auf eine Tendenz zu verlängertem Überleben mit einer Mehrfachkombinationstherapie hindeutet2).
Yang N, Kong D, Wang X, Liu Y. Perianal rhabdomyosarcoma in an adult: A case report and review of the literature. Medicine. 2023;102(48):e36199.
Chang T, Ding C, Liu Y, Yang Y, Mao Q.. Primary pineal alveolar rhabdomyosarcoma in an adult patient: a case report and literature review. BMC Neurol. 2025;25(1):106. doi:10.1186/s12883-025-04113-8. PMID:40082819; PMCID:PMC11905731.
Li Y, Li D, Wang J, Tang J.. Epithelioid and spindle rhabdomyosarcoma with TFCP2 rearrangement in abdominal wall: a distinctive entity with poor prognosis. Diagn Pathol. 2023;18(1):41. doi:10.1186/s13000-023-01330-y. PMID:36998041; PMCID:PMC10061849.
Kopieren Sie den Artikeltext und fügen Sie ihn in den KI-Assistenten Ihrer Wahl ein.
Artikel in die Zwischenablage kopiert
Öffnen Sie unten einen KI-Assistenten und fügen Sie den kopierten Text in den Chat ein.