Il rabdomiosarcoma è un tumore maligno derivante dalle cellule mesenchimali dell’orbita, che mostra differenziazione muscolare striata. Origina da cellule mesenchimali indifferenziate dell’orbita, non dalla trasformazione maligna di tessuto muscolare differenziato come i muscoli extraoculari. È il tumore maligno orbitale più frequente nei bambini; di fronte a un tumore orbitale a rapida crescita in un bambino, si deve pensare per primo a questa malattia.
Negli Stati Uniti, rappresenta circa il 5% di tutti i tumori pediatrici, con circa 250 nuovi casi diagnosticati ogni anno. Di questi, circa il 10% (circa 35 casi all’anno) sono di origine orbitale. L’incidenza annuale è stimata in 4 casi per milione di abitanti nella popolazione generale e 4,5 casi per milione di bambini1).
L’età media alla diagnosi è di 7-8 anni; due terzi dei casi si verificano prima dei 10 anni. Il 90% dei casi si verifica prima dei 16 anni, con una leggera predominanza maschile (rapporto 5:3). Non ci sono differenze di incidenza per razza. Il tumore è ben delimitato e spesso localizzato nella parte superiore dell’orbita.
La distribuzione delle sedi del rabdomiosarcoma oculare è: orbita 76%, congiuntiva 12%, uvea 9%, palpebra 3%. Può insorgere all’interno dell’orbita o provenire dai tessuti circostanti come i seni paranasali e infiltrare l’orbita. L’orbita è il sito primario del 10% di tutti i rabdomiosarcomi; altre sedi includono il tratto urogenitale (22%), gli arti (18%), le regioni parameningee (16%) e altre sedi della testa e del collo (10%).
QA quale età si verifica più frequentemente il rabdomiosarcoma?
A
Due terzi dei casi si verificano in bambini di età inferiore a 10 anni, con un’età media di 7-8 anni. Il 90% dei casi si verifica prima dei 16 anni. L’insorgenza negli adulti è rara ma riportata.
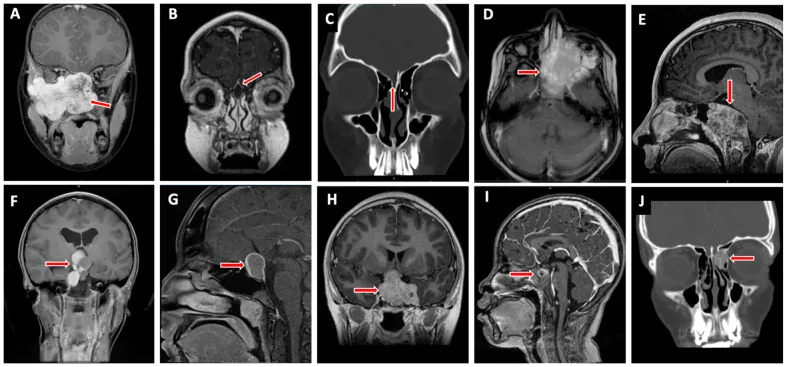
Valencia-Sanchez BA, et al. Special Considerations in Pediatric Endoscopic Skull Base Surgery. J Clin Med. 2024. Figure 2. PMCID: PMC11013018. License: CC BY.
La freccia rossa indica un rabdomiosarcoma (D), una delle lesioni tipiche della base cranica nei bambini. Ciò corrisponde all’infiltrazione tumorale trattata nella sezione «2. Principali sintomi e segni clinici».
Es oftalmo rapido: Un esoftalmo unilaterale che progredisce rapidamente in poche settimane è un sintomo tipico.
Gonfiore palpebrale: Con la crescita del tumore, la palpebra si gonfia. Anche in caso di gonfiore marcato, il rossore è lieve, i segni infiammatori sono scarsi e il dolore non è intenso, caratteristicamente.
Limitazione dei movimenti oculari, ptosi, ecc., che presentano vari quadri clinici a seconda della sede del tumore.
Si verifica frequentemente nei bambini di età 0-9 anni ed è caratterizzato da una rapida progressione.
Le lesioni sono più frequenti nelle regioni superiore e supero-interna, e la massa devia il bulbo oculare verso il basso e l’esterno. Si osserva una massa rotonda o ovale, con confini relativamente netti, al di fuori del cono muscolare.
Circa il 10% dei casi origina dai seni paranasali e dalla cavità nasale, invadendo secondariamente l’orbita, e può presentarsi con sinusite, ostruzione nasale o epistassi. I tumori posteriori possono associarsi a pieghe coroideali, distacco di retina o edema della papilla ottica.
Nei casi primitivi della congiuntiva, il tumore appare come una massa granulomatosa rosa a grappolo nel fornice congiuntivale. Nei casi primitivi dell’uvea, si presenta come una massa iridea e può associarsi a disseminazione in camera anteriore o glaucoma secondario.
Le metastasi sono più frequenti nei polmoni, seguite da midollo osseo, ossa e linfonodi. L’orbita è quasi priva di vasi linfatici, ma i tumori anteriori della congiuntiva e delle palpebre possono metastatizzare ai linfonodi regionali.
QQuali malattie possono essere confuse con il rabdomiosarcoma?
A
Le diagnosi differenziali cliniche includono neuroblastoma, cloroma, linfangioma, emangioma infantile, cellulite orbitaria e infiammazione aspecifica. Inoltre, l’estensione orbitaria di sinusite, rottura di cisti dermoide, sarcoma di Ewing e infiltrazione orbitaria leucemica sono incluse nella diagnosi differenziale. La differenziazione si basa sulla rapida progressione, sull’imaging e sui risultati della biopsia.
Il rabdomiosarcoma non origina dai muscoli extraoculari, ma da cellule mesenchimali indifferenziate pluripotenti nei tessuti molli orbitari. La maggior parte dei casi è sporadica e la causa esatta è sconosciuta.
I fattori di rischio genetici associati sono i seguenti:
Neurofibromatosi tipo 1: nota per essere associata a un aumentato rischio di rabdomiosarcoma.
Sindrome di Li-Fraumeni: coinvolge anomalie del gene soppressore tumorale p53.
Sindrome di Beckwith-Wiedemann: malattia congenita caratterizzata da crescita eccessiva, ernia ombelicale e ipoglicemia.
Retinoblastoma ereditario: può verificarsi come tumore secondario dopo radioterapia.
Una TC cranica e orbitaria d’urgenza viene eseguita per verificare la presenza di un tumore intraorbitario, la sua omogeneità, la distruzione della parete orbitaria e l’estensione intracranica o sinusale. Successivamente, viene eseguita una RM per valutare la natura interna del tumore e i suoi rapporti con il bulbo oculare e i muscoli extraoculari.
I reperti caratteristici di ciascuna modalità sono i seguenti:
Esame
Principali reperti
TC
Tumore rotondo-ovale, omogeneo, a margini netti. Enhancement moderato-marcato.
RM T1
Ipointenso rispetto al grasso orbitario, isointenso rispetto ai muscoli extraoculari.
Alla RM, la differenziazione dall’emangioma capillare può essere difficile, ma l’emangioma capillare è ricco di vasi (flusso sanguigno), consentendo la distinzione. Quando la componente stromale è abbondante, l’intensità del segnale RM è diversa.
La ricerca di metastasi comprende radiografia del torace, scintigrafia ossea e citologia su aspirato midollare. Per la ricerca sistemica si utilizzano anche PET/TC, TC total body e scintigrafia.
Per la diagnosi definitiva è necessaria una biopsia. L’agobiopsia aspirativa non è sufficiente; si esegue una biopsia escissionale o incisionale.
Biopsia escissionale: scelta quando è possibile la rimozione chirurgica senza danneggiare strutture importanti.
Biopsia incisionale: scelta quando il tumore è grande e situato nell’orbita posteriore.
L’esame estemporaneo intraoperatorio può non distinguere il tumore da altre neoplasie. Il principio è eseguire una biopsia in urgenza e iniziare precocemente chemioterapia e radioterapia.
La dimostrazione di rabdomioblasti mediante microscopia ottica, immunoistochimica e microscopia elettronica è il cardine della diagnosi.
Le striature vengono rilevate con colorazione HE o tricromica di Masson. Per la differenziazione delle cellule indifferenziate si esegue l’immunoistochimica; un pattern desmina positivo, HHF-35 (actina) positivo, α-actina muscolare liscia negativo è utile per la diagnosi. La microscopia elettronica mostra fasci di actina-miosina e proteine delle bande A, I e Z.
L’asportazione chirurgica completa non è possibile e non deve essere l’obiettivo. Attualmente, lo scopo principale della chirurgia è ottenere una diagnosi tissutale; l’epoca dell’exenteratio orbitae come prima scelta è finita. Dopo la conferma della diagnosi patologica mediante biopsia escissionale, in pediatria si eseguono TC toraco-addominale e biopsia midollare per verificare la presenza di metastasi a distanza, ad esempio polmonari. La chemioterapia sistemica è la base del trattamento; la terapia triplice che combina chirurgia, radioterapia e chemioterapia è il trattamento standard.
La chemioterapia utilizza principalmente il regime VAC (vincristina + actinomicina D + ciclofosfamide). Sono stati riportati anche benefici di ifosfamide ed etoposide.
La radioterapia viene somministrata con circa 40-60 Gy. Dall’aprile 2016, la protonterapia è coperta dall’assicurazione sanitaria e si è aggiunta come opzione terapeutica standard. La protonterapia ha il vantaggio di ridurre la dose ai tessuti normali. In alcuni casi viene eseguito anche il trapianto di cellule staminali ematopoietiche.
Il rabdomiosarcoma orbitario viene trattato in base alla classificazione dell’Intergroup Rhabdomyosarcoma Study (IRS). In una serie di 30 casi di Shields et al., la distribuzione era: gruppo I 7%, gruppo II 37%, gruppo III 53%, gruppo IV 3%.
Gruppo
Definizione
Strategia terapeutica
I
Resezione completa della malattia locale
Solo chemioterapia
II
Malattia residua o metastasi linfonodali regionali
Chemioterapia + radioterapia
III
Resezione incompleta o residuo macroscopico
Chemioterapia + radioterapia
IV
Metastasi a distanza
Chemioterapia + radioterapia
Per i gruppi II–IV, la radioterapia viene somministrata con 4000–5000 cGy (40–50 Gy) in 4–5 settimane.
Nei casi non resecabili o recidivanti, si considera la radioterapia palliativa o la chemioterapia. Il trattamento chirurgico può includere l’asportazione del tumore o l’exenteratio orbitae. Anche in caso di resezione completa, la chemioterapia è obbligatoria. L’orbita è un sito a prognosi favorevole; con un trattamento adeguato, la sopravvivenza attesa è superiore al 90%.
L’infiltrazione intracranica o dei seni paranasali, o le metastasi polmonari, sistemiche o ai linfonodi cervicali, comportano una prognosi infausta. La diagnosi precoce e l’inizio tempestivo della chemioterapia migliorano la risposta e la prognosi.
Le complicanze oftalmiche della chemioterapia includono cheratocongiuntivite secca e blefarocongiuntivite da ciclofosfamide, congiuntivite e visione offuscata da ifosfamide, e occlusione dell’arteria centrale della retina da etoposide.
Le recidive locali vengono valutate con risonanza magnetica regolare. Le metastasi ematogene sono frequenti, pertanto è necessario uno screening sistemico regolare.
Gli esiti visivi a lungo termine dopo il trattamento (Shields) sono: 20/20–20/40 nel 39%, 20/50–20/100 nel 18% e 20/200–nessuna percezione luminosa nel 43%.
QQuali complicanze oculari possono verificarsi dopo il trattamento del rabdomiosarcoma?
A
Le principali complicanze dopo radioterapia (4000–5000 cGy) includono retinopatia da radiazioni (90%), cataratta da radiazioni (55%), occhio secco (36%), ipoplasia orbitaria (24%) e ptosi (9%). Anche la chemioterapia può causare cheratocongiuntivite secca, congiuntivite o occlusione dell’arteria centrale della retina a seconda del farmaco.
Secondo la classificazione OMS, il rabdomiosarcoma è suddiviso in quattro sottotipi: embrionale, alveolare, pleomorfo e a cellule fusate/sclerosante 2).
Tipo embrionale
Frequenza: rappresenta il 50-60% di tutti i rabdomiosarcomi e l’80-84% dei rabdomiosarcomi orbitali.
Istologia: fasci di cellule fusiformi (cellule a forma di fuso) orientati in varie direzioni. Cellule piccole rotonde o fusiformi con scarso pleomorfismo nucleare. Citoplasma scarso con granuli eosinofili, e presenza di rabdomioblasti striati. Più frequente nella parte inferiore dell’orbita.
Prognosi: il tasso di sopravvivenza a 5 anni per i casi orbitali è del 95%, favorevole. Età media di insorgenza 7-8 anni.
Tipo alveolare
Frequenza: circa il 20% di tutti i rabdomiosarcomi. Circa il 10% dei rabdomiosarcomi orbitali.
Istologia: il tumore cresce in nidi alveolari. Grandi cellule irregolari con nucleo voluminoso e abbondante citoplasma eosinofilo, disposte attorno a trabecole fibrose. Tendenza a metastasi estese.
Prognosi: il tasso di sopravvivenza a 5 anni per i casi orbitali è del 74%. È il tipo istologico più maligno e con la prognosi peggiore.
Tipo pleomorfo (differenziato)
Frequenza: raro nei bambini. Si verifica principalmente negli arti degli adulti (soprattutto coscia).
Istologia: cellule multinucleate, da rotonde ad allungate, con abbondante citoplasma, in cui si possono identificare strie. Costituito da cellule tumorali altamente pleomorfe. Poco frequente ma altamente differenziato.
Prognosi: relativamente favorevole nei casi orbitali.
Il tipo botrioide (botryoid) è una variante del tipo embrionale frequente nei neonati, caratterizzata da aggregati di cellule tumorali subepiteliali che conferiscono un aspetto «a grappolo d’uva».
Sedi a prognosi favorevole: orbita, tratto urogenitale (eccetto vescica e prostata), testa e collo (eccetto parameningei)
Sedi a prognosi sfavorevole: arti, vescica, prostata, parameningei
Istologicamente, i tipi alveolare e pleomorfo hanno una prognosi peggiore rispetto al tipo embrionale. Il tasso di sopravvivenza a 5 anni per l’intero rabdomiosarcoma è del 66%, ma il rabdomiosarcoma orbitario è classificato nel gruppo a prognosi favorevole a causa della sua localizzazione.
Il rabdomiosarcoma alveolare è caratterizzato da traslocazioni cromosomiche ricorrenti t(2;13)(q35;q14) e t(1;13)(p36;q14). Circa l’80% dei rabdomiosarcomi alveolari coinvolge traslocazioni PAX3-FOXO1 o PAX7-FOXO1. La fusione PAX3-FOXO1 è associata a un’alta espressione di OLIG2 e a una prognosi sfavorevole2).
Il rabdomiosarcoma embrionale non presenta riarrangiamenti cromosomici strutturali ricorrenti, ma si osserva frequentemente una perdita allelica sul cromosoma 11 (in particolare la regione 11p15.5).
QQuali sono le differenze prognostiche tra il tipo embrionale e quello alveolare?
A
Nel rabdomiosarcoma orbitario, il tasso di sopravvivenza a 5 anni per il tipo embrionale è del 95%, mentre per il tipo alveolare è inferiore, pari al 74%. Il tipo alveolare è associato a traslocazioni cromosomiche come PAX3-FOXO1 ed è un tipo istologico soggetto a metastasi estese.
7. Ricerche recenti e prospettive future (rapporti in fase di ricerca)
Recentemente è stato identificato un nuovo sottotipo chiamato rabdomiosarcoma a cellule fusiformi epitelioidi con riarrangiamento di TFCP2.
Li et al. (2023) hanno riportato che il rabdomiosarcoma a cellule fusiformi epitelioidi si verifica frequentemente nelle ossa (testa e collo, bacino) ed è caratterizzato da fusioni EWSR1-TFCP2 o FUS-TFCP2, un sottotipo con prognosi estremamente sfavorevole3). La sopravvivenza mediana riportata è di soli 17 mesi.
La ricerca su terapie mirate alla proteina di fusione PAX3-FOXO1, coinvolta in circa l’80% dei rabdomiosarcomi alveolari, è in corso.
È stato riportato che un preparato contenente piccoli RNA interferenti (siRNA) incapsulati in particelle liposoma-protamina regola efficacemente verso il basso l’espressione di PAX3-FOXO1 in linee cellulari di rabdomiosarcoma alveolare in vitro e provoca ritardo e soppressione della crescita di tumori da xenotrapianto di rabdomiosarcoma alveolare1).
L’applicazione di inibitori dei checkpoint immunitari (come nivolumab) al sarcoma metastatico è in fase di sperimentazione1). Tuttavia, al momento la loro efficacia contro il rabdomiosarcoma non è stabilita.
Trattamento del rabdomiosarcoma primitivo dell’adulto
Una revisione della letteratura sul rabdomiosarcoma alveolare primitivo della regione pineale nell’adulto suggerisce un’associazione tra intensità del trattamento e sopravvivenza.
In una revisione di 13 casi di rabdomiosarcoma alveolare primitivo della regione pineale nell’adulto di Chang et al. (2025), la sopravvivenza media era di circa 5 mesi con la sola chirurgia, di circa 10,28 mesi con chirurgia più radioterapia e di circa 11,33 mesi con chirurgia più radioterapia più chemioterapia, mostrando una tendenza a una sopravvivenza prolungata con il trattamento combinato multimodale2).
Yang N, Kong D, Wang X, Liu Y. Perianal rhabdomyosarcoma in an adult: A case report and review of the literature. Medicine. 2023;102(48):e36199.
Chang T, Ding C, Liu Y, Yang Y, Mao Q.. Primary pineal alveolar rhabdomyosarcoma in an adult patient: a case report and literature review. BMC Neurol. 2025;25(1):106. doi:10.1186/s12883-025-04113-8. PMID:40082819; PMCID:PMC11905731.
Li Y, Li D, Wang J, Tang J.. Epithelioid and spindle rhabdomyosarcoma with TFCP2 rearrangement in abdominal wall: a distinctive entity with poor prognosis. Diagn Pathol. 2023;18(1):41. doi:10.1186/s13000-023-01330-y. PMID:36998041; PMCID:PMC10061849.
Copia il testo dell'articolo e incollalo nell'assistente IA che preferisci.
Articolo copiato negli appunti
Apri un assistente IA qui sotto e incolla il testo copiato nella chat.