O rabdomiossarcoma é um tumor maligno derivado de células mesenquimais da órbita, mostrando diferenciação para músculo estriado. Origina-se de células mesenquimais indiferenciadas dentro da órbita, e não de tecido muscular diferenciado, como os músculos extraoculares. É o tumor maligno orbitário mais frequente em crianças e deve ser a primeira consideração ao se deparar com um tumor orbitário de rápida expansão em uma criança.
Nos Estados Unidos, representa cerca de 5% de todos os cânceres infantis, com aproximadamente 250 novos casos diagnosticados anualmente. Cerca de 10% deles (cerca de 35 casos por ano) são primários da órbita. A incidência anual é estimada em 4 casos por milhão de habitantes na população geral e 4,5 casos por milhão de crianças1).
A idade média ao diagnóstico é de 7-8 anos, e dois terços ocorrem em crianças menores de 10 anos. 90% ocorrem antes dos 16 anos, com ligeiro predomínio no sexo masculino (proporção 5:3). Não há diferença de incidência por raça. O tumor é bem delimitado e ocorre mais frequentemente na parte superior da órbita.
A distribuição dos locais do rabdomiossarcoma ocular é: órbita 76%, conjuntiva 12%, úvea 9% e pálpebra 3%. Pode surgir dentro da órbita ou invadir a órbita a partir de tecidos circundantes, como os seios paranasais. A órbita é o local primário em 10% de todos os rabdomiossarcomas; outros locais incluem trato urogenital (22%), extremidades (18%), região parameníngea (16%) e outros locais de cabeça e pescoço (10%).
QEm que idade o rabdomiossarcoma ocorre com mais frequência?
A
2/3 dos casos ocorrem em crianças com menos de 10 anos, com idade média de início de 7-8 anos. 90% ocorrem antes dos 16 anos. É raro em adultos, mas já foi relatado.
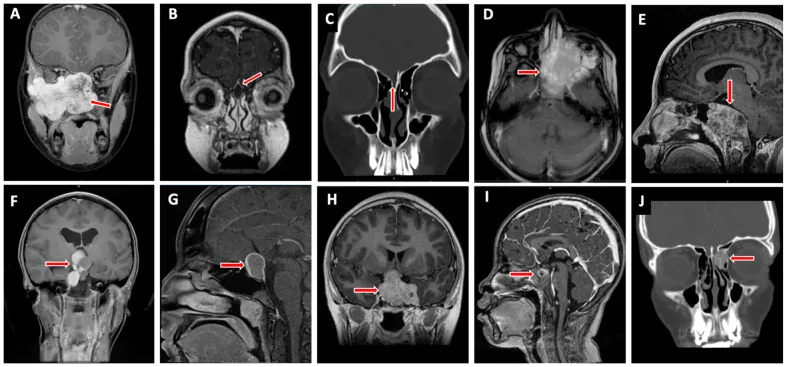
Valencia-Sanchez BA, et al. Special Considerations in Pediatric Endoscopic Skull Base Surgery. J Clin Med. 2024. Figure 2. PMCID: PMC11013018. License: CC BY.
A seta vermelha indica rabdomiossarcoma (D), uma das lesões típicas na base do crânio em crianças. Corresponde à infiltração tumoral discutida na seção “2. Principais sintomas e achados clínicos”.
Proptose rápida: Proptose unilateral que progride rapidamente em semanas é um sintoma típico.
Inchaço palpebral: A pálpebra incha conforme o tumor cresce. Mesmo com inchaço intenso, o rubor é leve e os sinais inflamatórios são escassos, a dor não é intensa, o que é característico.
Restrição dos movimentos oculares, ptose e várias outras apresentações clínicas dependendo da localização do tumor.
Ocorre comumente em crianças de 0-9 anos e é caracterizado por progressão rápida.
A lesão é mais comum na região superior ou súpero-medial, e a massa desvia o globo ocular para baixo ou para fora. Observa-se uma massa redonda a oval, com bordas relativamente nítidas, fora do cone muscular.
Cerca de 10% dos casos originam-se dos seios paranasais ou cavidade nasal e invadem a órbita secundariamente, podendo apresentar sinusite, congestão nasal e epistaxe. Tumores posteriores podem estar associados a dobras coroidianas, descolamento de retina e edema de papila do nervo óptico.
Nos casos primários da conjuntiva, surge como uma massa granulomatosa rosada em forma de cacho de uva no fórnice conjuntival. Nos casos primários da úvea, surge como uma massa iriana e pode estar associada a semeadura na câmara anterior ou glaucoma secundário.
As metástases são mais frequentes nos pulmões, seguidas pela medula óssea, ossos e linfonodos. A órbita quase não possui vasos linfáticos, mas tumores anteriores da conjuntiva e pálpebra podem metastatizar para linfonodos regionais.
QQuais doenças podem ser confundidas com rabdomiossarcoma?
A
Os diagnósticos diferenciais clínicos incluem: neuroblastoma, cloroma, linfangioma, hemangioma infantil, celulite orbitária e inflamação inespecífica. Além disso, a extensão orbitária de sinusite, ruptura de cisto dermoide, sarcoma de Ewing e infiltração orbitária por leucemia também estão incluídos no diagnóstico diferencial. A diferenciação é feita com base na progressão rápida e nos achados de imagem e biópsia.
O rabdomiossarcoma origina-se de células mesenquimais indiferenciadas com potencial multipotente nos tecidos moles da órbita, e não dos músculos extraoculares. A maioria dos casos é esporádica e a causa exata é desconhecida.
Os fatores de risco genéticos associados são os seguintes:
Neurofibromatose tipo 1: Conhecida por estar associada ao aumento do risco de rabdomiossarcoma.
Síndrome de Li-Fraumeni: Envolve anormalidade no gene supressor tumoral p53.
Síndrome de Beckwith-Wiedemann: Distúrbio congênito caracterizado por crescimento excessivo, hérnia umbilical e hipoglicemia.
Retinoblastoma hereditário: Pode ocorrer como tumor secundário após radioterapia.
A tomografia computadorizada (TC) de crânio e órbita é realizada com urgência para verificar a presença de tumor na órbita, homogeneidade do tumor, destruição da parede óssea orbitária e extensão intracraniana ou para os seios paranasais. Em seguida, a ressonância magnética (RM) é realizada para avaliar as características internas do tumor e sua relação com o globo ocular e os músculos extraoculares.
Os achados característicos de cada modalidade são os seguintes:
Exame
Principais Achados
TC
Massa homogênea, bem delimitada, redonda a oval. Realce de contraste moderado a intenso
RM T1
Hipossinal em relação à gordura orbitária, isossinal em relação aos músculos extraoculares
Na RM, às vezes é difícil diferenciar do hemangioma capilar, mas o hemangioma capilar é rico em vasos (fluxo sanguíneo), permitindo a distinção. Se houver muitos componentes intersticiais, a intensidade do sinal na RM é diferente.
Para pesquisa de metástases, realiza-se radiografia de tórax, cintilografia óssea e citologia de aspirado de medula óssea. Para busca sistêmica, também são usados PET/TC, TC de corpo inteiro e cintilografia.
Para o diagnóstico definitivo, é necessária biópsia. A biópsia aspirativa por agulha fina é insuficiente; realiza-se biópsia excisional ou incisional.
Biópsia excisional: Escolhida quando o tumor pode ser removido cirurgicamente sem danificar estruturas importantes.
Biópsia incisional: Escolhida quando o tumor é grande e localizado na órbita posterior.
No diagnóstico intraoperatório rápido, às vezes é difícil diferenciar de outros tumores. O princípio é realizar biópsia em cirurgia de urgência e iniciar quimioterapia e radioterapia precocemente.
O diagnóstico baseia-se na demonstração de rabdomioblastos por microscopia óptica, imuno-histoquímica e microscopia eletrônica.
As estrias são detectadas pela coloração HE e tricrômico de Masson. Para diferenciar células indiferenciadas, realiza-se imuno-histoquímica, sendo o padrão desmina positivo, HHF-35 (actina) positivo e α-actina de músculo liso negativo útil para o diagnóstico. A microscopia eletrônica revela feixes de actina-miosina e proteínas das bandas A, I e Z.
Doenças de diferenciação clínica: neuroblastoma, cloroma, linfangioma, hemangioma infantil, celulite orbitária, inflamação inespecífica. Além disso, extensão de sinusite para a órbita, celulite orbitária, ruptura de cisto dermoide, hemorragia intratumoral de hemangioma, hemorragia de linfangioma, sarcoma de Ewing, infiltração leucêmica na órbita (cloroma) e hemorragia orbitária também estão incluídas no diagnóstico diferencial.
A excisão cirúrgica total não é possível e não deve ser o objetivo. Atualmente, o principal objetivo da cirurgia é obter o diagnóstico tecidual, e a era da exenteração orbitária como primeira escolha terminou. Após a confirmação do diagnóstico patológico por biópsia excisional, são realizados TC de tórax e abdome e biópsia de medula óssea no departamento de pediatria para verificar metástases pulmonares ou sistêmicas. A quimioterapia sistêmica é a base do tratamento, e a terapia combinada de três modalidades (cirurgia, radioterapia e quimioterapia) é o tratamento padrão.
Na quimioterapia, o regime VAC (vincristina + actinomicina D + ciclofosfamida) é usado principalmente. Além disso, o benefício de ifosfamida e etoposídeo também foi relatado.
A radioterapia é administrada com cerca de 40-60 Gy. A partir de abril de 2016, a terapia com prótons passou a ser coberta pelo seguro saúde e foi adicionada como opção de tratamento padrão. A terapia com prótons tem a vantagem de reduzir a dose nos tecidos normais. O transplante de células-tronco hematopoéticas também é realizado em alguns casos.
O plano de tratamento para rabdomiossarcoma orbitário é determinado com base na classificação do Intergroup Rhabdomyosarcoma Study (IRS). Em 30 casos de Shields et al., a distribuição dos grupos foi: Grupo I 7%, Grupo II 37%, Grupo III 53%, Grupo IV 3%.
Grupo
Definição
Plano de Tratamento
I
Ressecção completa da doença local
Apenas quimioterapia
II
Doença residual ou metástase linfonodal regional
Quimioterapia + Radioterapia
III
Ressecção incompleta ou residual macroscópico
Quimioterapia + Radioterapia
IV
Metástase à distância
Quimioterapia + Radioterapia
A radioterapia para os grupos II a IV é de 4000-5000 cGy (40-50 Gy) ao longo de 4-5 semanas.
Em casos não ressecáveis totalmente ou recidivantes, considere radioterapia ou quimioterapia como tratamento paliativo. O tratamento cirúrgico pode incluir excisão tumoral ou exenteração orbitária. Mesmo com ressecção completa, a quimioterapia é obrigatória. A órbita é um local de bom prognóstico; com tratamento adequado, pode-se esperar sobrevida acima de 90%.
Se houver invasão intracraniana ou dos seios paranasais, ou metástase para pulmões ou linfonodos cervicais, o prognóstico de vida é ruim. A detecção precoce e o início precoce da quimioterapia resultam em boa resposta à quimioterapia e bom prognóstico de vida.
Complicações oftalmológicas da quimioterapia incluem ceratoconjuntivite seca e blefaroconjuntivite por ciclofosfamida, conjuntivite e visão turva por ifosfamida, e oclusão da artéria central da retina por etoposídeo, conforme relatado.
A recidiva local é avaliada por meio de exames de ressonância magnética regulares. Metástases hematogênicas são frequentes, portanto exames sistêmicos regulares são realizados.
Os resultados visuais de longo prazo após o tratamento (Shields) são: 20/20 a 20/40 em 39%, 20/50 a 20/100 em 18% e 20/200 a ausência de percepção de luz em 43%.
QQuais complicações oculares podem ocorrer após o tratamento do rabdomiossarcoma?
O rabdomiossarcoma é classificado pela OMS em 4 subtipos: embrionário, alveolar, pleomórfico e de células fusiformes/esclerosante 2).
Tipo embrionário
Frequência: Representa 50-60% de todos os rabdomiossarcomas e 80-84% dos rabdomiossarcomas orbitários.
Histologia: Feixes de células fusiformes (células vilosas) correndo em várias direções. Células pequenas redondas ou fusiformes, com pouco pleomorfismo nuclear. Citoplasma escasso com grânulos eosinofílicos, e rabdomioblastos estriados são observados. Mais comum na parte inferior da órbita.
Prognóstico: A taxa de sobrevida em 5 anos para casos orbitários é de 95%, boa. Idade média de início: 7-8 anos.
Tipo Alveolar
Frequência: Cerca de 20% de todos os rabdomiossarcomas. Cerca de 10% dos rabdomiossarcomas orbitários.
Histologia: O tumor cresce em padrão alveolar. Células grandes e irregulares com núcleos grandes e citoplasma rico corado por eosina, dispostas ao redor de trabéculas. Propenso a metástases extensas.
Prognóstico: A taxa de sobrevida em 5 anos para casos orbitários é de 74%. Este é o tipo histológico de maior malignidade e pior prognóstico.
Tipo Pleomórfico (Diferenciado)
Frequência: Raro em crianças. Ocorre principalmente em extremidades de adultos (especialmente coxa).
Histologia: Células redondas a alongadas, multinucleadas, com citoplasma rico, estrias identificáveis. Composto por células tumorais altamente pleomórficas. Baixa frequência, mas alto grau de diferenciação.
Prognóstico: Relativamente bom em casos orbitários.
O tipo Botrioide (botryoid) é uma variante do tipo embrionário comum em lactentes, caracterizado por agregados de células tumorais subepiteliais que conferem uma aparência “semelhante a uvas”.
Locais de bom prognóstico: Órbita, trato urogenital exceto bexiga e próstata, cabeça e pescoço exceto parameninge
Locais de mau prognóstico: Extremidades, bexiga, próstata, parameninge
Histologicamente, o prognóstico dos tipos alveolar e pleomórfico é pior em comparação ao tipo embrionário. A taxa de sobrevida em 5 anos para o rabdomiossarcoma como um todo é de 66%, mas o rabdomiossarcoma orbitário é classificado como de bom prognóstico com base na localização.
O rabdomiossarcoma alveolar é caracterizado por translocações cromossômicas recorrentes t(2;13)(q35;q14) e t(1;13)(p36;q14). Cerca de 80% dos rabdomiossarcomas alveolares envolvem translocações PAX3-FOXO1 ou PAX7-FOXO1. A fusão PAX3-FOXO1 está associada à alta expressão de OLIG2 e mau prognóstico2).
O rabdomiossarcoma embrionário não apresenta rearranjos estruturais cromossômicos recorrentes, mas a perda alélica no cromossomo 11 (especialmente a região 11p15.5) é frequentemente observada.
QQual a diferença no prognóstico entre os tipos embrionário e alveolar?
A
A taxa de sobrevida em 5 anos para o tipo embrionário no rabdomiossarcoma orbitário é de 95%, enquanto para o tipo alveolar é de 74%, mais baixa. O tipo alveolar é acompanhado por translocações cromossômicas como PAX3-FOXO1 e é um tipo histológico propenso a metástases extensas.
7. Pesquisas recentes e perspectivas futuras (relatos em fase de pesquisa)
Nos últimos anos, um novo subtipo denominado rabdomiossarcoma de células fusiformes epitelioides com rearranjo de TFCP2 foi identificado.
Li et al. (2023) relataram que o rabdomiossarcoma de células fusiformes epitelioides ocorre frequentemente em ossos (cabeça e pescoço, pelve) e é caracterizado por fusões EWSR1-TFCP2 ou FUS-TFCP2, um subtipo de prognóstico extremamente ruim3). A mediana de sobrevida dos casos relatados é de apenas 17 meses.
Pesquisas terapêuticas visando a proteína de fusão PAX3-FOXO1, envolvida em cerca de 80% dos rabdomiossarcomas alveolares, estão em andamento.
Foi relatado que uma formulação contendo pequeno RNA interferente (siRNA) encapsulado em partículas de lipossoma-protamina regula negativamente a expressão de PAX3-FOXO1 de forma eficiente em células de rabdomiossarcoma alveolar in vitro, e causa atraso ou supressão do crescimento de tumores de xenoenxerto de rabdomiossarcoma alveolar1).
Inibidores de checkpoint imune (como nivolumabe) estão sendo testados em sarcomas metastáticos1). No entanto, a eficácia no rabdomiossarcoma ainda não foi estabelecida no momento.
Tratamento do Rabdomiossarcoma Primário em Adultos
Em uma revisão de literatura sobre rabdomiossarcoma alveolar primário da região da glândula pineal em adultos, foi sugerida uma associação entre a intensidade do tratamento e a sobrevida.
Em uma revisão de Chang et al. (2025) de 13 casos de rabdomiossarcoma alveolar primário da glândula pineal em adultos, foi relatado que a sobrevida média com cirurgia isolada foi de aproximadamente 5 meses, com cirurgia + radioterapia foi de aproximadamente 10,28 meses, e com cirurgia + radioterapia + quimioterapia foi de aproximadamente 11,33 meses, indicando que o tratamento combinado prolonga a sobrevida2).
Yang N, Kong D, Wang X, Liu Y. Perianal rhabdomyosarcoma in an adult: A case report and review of the literature. Medicine. 2023;102(48):e36199.
Chang T, Ding C, Liu Y, Yang Y, Mao Q.. Primary pineal alveolar rhabdomyosarcoma in an adult patient: a case report and literature review. BMC Neurol. 2025;25(1):106. doi:10.1186/s12883-025-04113-8. PMID:40082819; PMCID:PMC11905731.
Li Y, Li D, Wang J, Tang J.. Epithelioid and spindle rhabdomyosarcoma with TFCP2 rearrangement in abdominal wall: a distinctive entity with poor prognosis. Diagn Pathol. 2023;18(1):41. doi:10.1186/s13000-023-01330-y. PMID:36998041; PMCID:PMC10061849.
Copie o texto do artigo e cole no assistente de IA de sua preferência.
Artigo copiado para a área de transferência
Abra um assistente de IA abaixo e cole o texto copiado na conversa.