رابدومیوسارکوم یک تومور بدخیم است که از سلولهای مزانشیمی اربیت منشأ گرفته و تمایز به عضله مخطط را نشان میدهد. از سلولهای مزانشیمی تمایزنیافته درون اربیت ایجاد میشود و ناشی از بدخیمی بافت عضلانی تمایز یافته مانند عضلات خارج چشمی نیست. این شایعترین تومور بدخیم اربیت در کودکان است و در صورت مشاهده تومور اربیت با رشد سریع در کودک، باید ابتدا به آن فکر کرد.
در ایالات متحده، حدود ۵٪ از کل سرطانهای کودکان را تشکیل میدهد و سالانه حدود ۲۵۰ مورد جدید تشخیص داده میشود. از این تعداد، حدود ۱۰٪ (حدود ۳۵ مورد در سال) منشأ اربیتی دارند. بروز سالانه در کل جمعیت ۴ مورد در هر میلیون نفر و در کودکان ۴.۵ مورد در هر میلیون نفر تخمین زده میشود1).
میانگین سن بروز ۷ تا ۸ سال است و دو سوم موارد در کودکان زیر ۱۰ سال رخ میدهد. ۹۰٪ موارد در سنین زیر ۱۶ سال بروز میکند و در پسران کمی شایعتر است (نسبت پسر به دختر ۵:۳). تفاوت نژادی در میزان بروز وجود ندارد. تومور معمولاً با حدود مشخص بوده و در قسمت فوقانی اربیت شایعتر است.
توزیع محل بروز رابدومیوسارکوم چشمی به این صورت است: اربیت ۷۶٪، ملتحمه ۱۲٪، یووه آ ۹٪، پلک ۳٪. ممکن است درون اربیت ایجاد شود یا توموری که در بافتهای اطراف مانند سینوسهای پارانازال ایجاد شده به اربیت نفوذ کند. اربیت محل اولیه ۱۰٪ از کل رابدومیوسارکومها است و سایر محلهای بروز شامل دستگاه ادراری-تناسلی (۲۲٪)، اندامها (۱۸٪)، نزدیک مننژ (۱۶٪) و سایر نواحی سر و گردن (۱۰٪) است.
Qرابدومیوسارکوم در چه سنی بیشتر بروز میکند؟
A
دو سوم موارد در کودکان زیر 10 سال رخ میدهد و میانگین سن شروع 7 تا 8 سال است. 90% موارد در سنین زیر 16 سال رخ میدهد. بروز در بزرگسالان نادر است اما گزارش شده است.
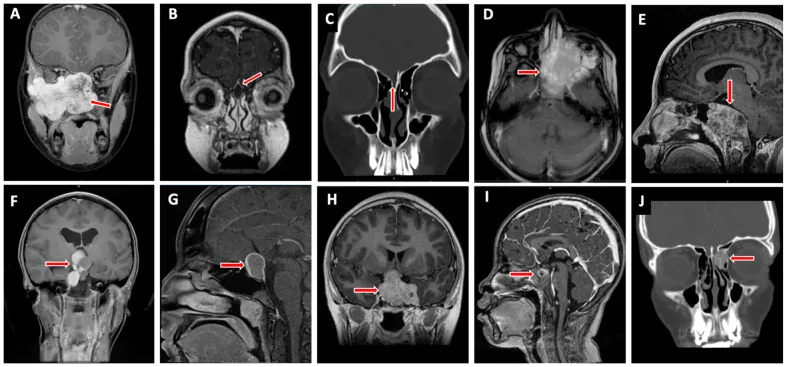
Valencia-Sanchez BA, et al. Special Considerations in Pediatric Endoscopic Skull Base Surgery. J Clin Med. 2024. Figure 2. PMCID: PMC11013018. License: CC BY.
فلش قرمز یکی از ضایعات شایع قاعده جمجمه در کودکان یعنی رابدومیوسارکوم (D) را نشان میدهد. این تصویر مربوط به نفوذ تومور است که در بخش «2. علائم اصلی و یافتههای بالینی» بحث شده است.
فراوانی یافتهها بر اساس گزارش Shields و همکاران به شرح زیر است:
یافته
فراوانی
برجستگی چشم
80 تا 100%
انحراف چشم
۸۰٪
تورم ملتحمه و پلک
۶۰٪
افتادگی پلک
۳۰ تا ۵۰٪
توده قابل لمس
۲۵٪
درد
۱۰٪
ضایعه اغلب در قسمت فوقانی یا فوقانی-داخلی قرار دارد و توده باعث جابجایی چشم به سمت پایین و خارج میشود. تودهای گرد تا بیضی با مرز نسبتاً مشخص در خارج از مخروط عضلانی دیده میشود.
حدود ۱۰٪ موارد از سینوسهای پارانازال و حفره بینی منشأ گرفته و ثانویه به مدار چشم حمله میکنند که ممکن است با سینوزیت، گرفتگی بینی و خونریزی بینی همراه باشد. تومورهای خلفی ممکن است با چینهای مشیمیه، جداشدگی شبکیه و ادم پاپی همراه باشند.
در موارد اولیه ملتحمه، تودهای دانهای صورتی و خوشهای مانند انگور در فورنیکس ملتحمه ظاهر میشود. در موارد اولیه یووه، به صورت توده عنبیه ظاهر شده و ممکن است با انتشار در اتاق قدامی و گلوکوم ثانویه همراه باشد.
متاستازها بیشتر در ریهها و سپس به ترتیب در مغز استخوان، استخوان و غدد لنفاوی دیده میشوند. اگرچه مدار چشم تقریباً فاقد عروق لنفاوی است، تومورهای قدامی ملتحمه و پلک میتوانند به غدد لنفاوی منطقهای متاستاز دهند.
Qچه بیماریهایی ممکن است با رابدومیوسارکوم اشتباه گرفته شوند؟
A
تشخیصهای افتراقی بالینی شامل نوروبلاستوما، کلروما، لنفانژیوم، همانژیوم نوزادی، سلولیت اربیت و التهاب غیراختصاصی است. همچنین گسترش سینوزیت به اربیت، پارگی درموئید کیست، سارکوم یوئینگ و نفوذ لوسمی به اربیت نیز در تشخیص افتراقی قرار میگیرند. با پیشرفت سریع و یافتههای تصویربرداری و بیوپسی افتراق داده میشود.
رابدومیوسارکوم از عضلات خارج چشمی منشأ نمیگیرد، بلکه از سلولهای مزانشیمی تمایزنیافته با توانایی تمایز چندگانه در بافت نرم اربیت ایجاد میشود. بیشتر موارد پراکنده هستند و علت مشخصی ندارند.
سیتی اسکن فوری سر و اربیت انجام میشود تا وجود تومور در اربیت، همگن بودن داخل تومور، تخریب دیواره استخوانی اربیت و گسترش به داخل جمجمه و سینوسها بررسی شود. سپس امآرآی برای تعیین ویژگیهای داخل تومور و رابطه موقعیتی با کره چشم و عضلات خارج چشمی انجام میشود.
یافتههای مشخصه هر روش به شرح زیر است:
روش
یافتههای اصلی
سیتی (CT)
توده گرد تا بیضی شکل، همگن و با مرز مشخص. افزایش کنتراست متوسط تا شدید
MRI T1
سیگنال کم نسبت به چربی کاسه چشم، سیگنال مساوی نسبت به عضلات خارج چشمی
MRI T2
سیگنال زیاد نسبت به چربی کاسه چشم و عضلات خارج چشمی
در MRI، گاهی افتراق از همانژیوم مویرگی دشوار است، اما همانژیوم مویرگی عروق (جریان خون) فراوانی دارد که امکان تمایز را فراهم میکند. اگر اجزای بینابینی زیاد باشند، شدت سیگنال MRI متفاوت است.
برای جستجوی متاستاز، رادیوگرافی قفسه سینه، سینتیگرافی استخوان و سیتولوژی آسپیراسیون مغز استخوان انجام میشود. برای بررسی سیستمیک، PET/CT، CT کل بدن و سینتیگرافی نیز استفاده میشود.
برای تشخیص قطعی، بیوپسی لازم است. آسپیراسیون با سوزن ظریف کافی نیست و بیوپسی اکسیژنال یا اینسیژنال انجام میشود.
بیوپسی اکسیژنال: زمانی انتخاب میشود که برداشت جراحی بدون آسیب به ساختارهای حیاتی امکانپذیر باشد.
بیوپسی اینسیژنال: زمانی انتخاب میشود که توده بزرگ و در قسمت خلفی کاسه چشم قرار دارد.
در تشخیص سریع حین عمل، گاهی افتراق از سایر تومورها دشوار است. اصل بر این است که بیوپسی به صورت اورژانسی انجام شود و شیمیدرمانی و رادیوتراپی زودهنگام آغاز گردد.
تشخیص بر اساس اثبات رابدومیوبلاستها با میکروسکوپ نوری، ایمونوهیستوشیمی و میکروسکوپ الکترونی است.
راههای عرضی با رنگآمیزی HE و Masson trichrome شناسایی میشوند. برای افتراق سلولهای تمایزنیافته، رنگآمیزی ایمونوهیستوشیمی انجام میشود و الگوی دسمین مثبت، HHF-35 (اکتین) مثبت و α-اکتین عضله صاف منفی برای تشخیص مفید است. در میکروسکوپ الکترونی، دستههای اکتین-میوزین و پروتئینهای نوارهای A، I و Z مشاهده میشوند.
بیماریهای بالینی افتراقی: نوروبلاستوم، کلروما، لنفانژیوم، همانژیوم نوزادی، سلولیت اربیت، التهاب غیراختصاصی. همچنین گسترش سینوزیت به اربیت، سلولیت اربیتال، پارگی کیست درموئید، خونریزی داخل تومور همانژیوم، خونریزی لنفانژیوم، سارکوم یوئینگ، نفوذ لوسمی به اربیت (کلروما) و خونریزی اربیت نیز در تشخیص افتراقی مطرح میشوند.
برداشت کامل تومور با جراحی امکانپذیر نیست و نباید هدف قرار گیرد. در حال حاضر، هدف اصلی جراحی تأمین تشخیص بافتی است و دوران انتخاب اولیه تخلیه محتویات اربیت به پایان رسیده است. پس از تأیید تشخیص پاتولوژیک با بیوپسی اکسیزیونال، در بخش اطفال سیتی اسکن قفسه سینه و شکم و بیوپسی مغز استخوان برای بررسی متاستازهای دوردست مانند ریه انجام میشود. شیمیدرمانی سیستمیک اساس درمان است و درمان ترکیبی شامل جراحی، پرتودرمانی و شیمیدرمانی درمان استاندارد محسوب میشود.
در شیمیدرمانی، رژیم VAC (وینکریستین + اکتینومایسین D + سیکلوفسفامید) عمدتاً استفاده میشود. همچنین فواید ایفوسفامید و اتوپوزید نیز گزارش شده است.
پرتودرمانی با دوز حدود 40 تا 60 گری انجام میشود. از آوریل 2016، پروتونتراپی تحت پوشش بیمه قرار گرفته است و به عنوان گزینهای در درمان استاندارد اضافه شده است. پروتونتراپی مزیت کاهش دوز به بافتهای سالم را دارد. پیوند سلولهای بنیادی خونساز نیز در برخی موارد انجام میشود.
رابدومیوسارکوم اربیت بر اساس طبقهبندی مرحلهای Intergroup Rhabdomyosarcoma Study (IRS) درمان میشود. در 30 مورد از Shields و همکاران، توزیع مراحل به صورت گروه I 7%، گروه II 37%، گروه III 53% و گروه IV 3% بود.
گروه
تعریف
رویکرد درمانی
I
برداشت کامل بیماری موضعی
فقط شیمیدرمانی
II
بیماری باقیمانده یا متاستاز به غدد لنفاوی ناحیهای
شیمیدرمانی + پرتودرمانی
III
برداشت ناقص یا باقیماندن تومور قابل مشاهده
شیمیدرمانی + پرتودرمانی
IV
متاستاز دوردست
شیمیدرمانی + پرتودرمانی
پرتودرمانی برای گروههای II تا IV به مدت ۴ تا ۵ هفته با دوز ۴۰۰۰ تا ۵۰۰۰ سانتیگری (۴۰ تا ۵۰ گری) انجام میشود.
در مواردی که برداشت کامل امکانپذیر نیست یا عود رخ میدهد، پرتودرمانی یا شیمیدرمانی به عنوان درمان تسکینی در نظر گرفته میشود. درمان جراحی شامل برداشت تومور و تخلیه محتویات حدقه نیز ممکن است انجام شود. حتی اگر تومور با جراحی به طور کامل برداشته شود، شیمیدرمانی ضروری است. حدقه ناحیهای با پیشآگهی خوب است و با درمان مناسب، بقای بیش از ۹۰٪ انتظار میرود.
اگر تومور به داخل جمجمه یا سینوسها نفوذ کرده باشد یا به ریهها و سایر نقاط بدن یا غدد لنفاوی گردن متاستاز داده باشد، پیشآگهی حیاتی بد است. تشخیص زودهنگام و شروع زودهنگام شیمیدرمانی، پاسخ به درمان و پیشآگهی حیاتی را بهبود میبخشد.
عوارض چشمی شیمیدرمانی شامل کراتوکونژونکتیویت خشک و بلفاروکونژونکتیویت ناشی از سیکلوفسفامید، کونژونکتیویت و تاری دید ناشی از ایفوسفامید، و انسداد شریان مرکزی شبکیه ناشی از اتوپوزید گزارش شده است.
عود موضعی با انجام منظم MRI ارزیابی میشود. متاستاز هماتوژن شایع است و معاینات سیستماتیک منظم انجام میشود.
نتایج بینایی بلندمدت پس از درمان (Shields): 20/20 تا 20/40 در 39٪، 20/50 تا 20/100 در 18٪، و 20/200 تا عدم درک نور در 43٪.
Qپس از درمان رابدومیوسارکوم چه عوارض چشمی ممکن است رخ دهد؟
A
عوارض اصلی پس از پرتودرمانی (4000 تا 5000 سانتیگری) شامل رتینوپاتی ناشی از پرتو (90٪)، آب مروارید ناشی از پرتو (55٪)، خشکی چشم (36٪)، هیپوپلازی حدقه (24٪) و پتوز پلک (9٪) گزارش شده است. شیمیدرمانی نیز بسته به دارو میتواند باعث کراتوکونژونکتیویت خشک، کونژونکتیویت و انسداد شریان مرکزی شبکیه شود.
رابدومیوسارکوم بر اساس طبقهبندی WHO به چهار زیرگروه جنینی، آلوئولار، پلئومورفیک و سلول دوکی/اسکلروزان تقسیم میشود2).
نوع جنینی
فراوانی: 50-60٪ از کل رابدومیوسارکومها و 80-84٪ از رابدومیوسارکومهای اربیت را تشکیل میدهد.
تصویر بافتی: دستههایی از سلولهای دوکی شکل (سلولهای پرزمانند) که در جهات مختلف قرار گرفتهاند. سلولها کوچک گرد یا دوکی شکل با پلئومورفیسم هستهای کم هستند. سیتوپلاسم کم و دارای گرانولهای اسیدوفیل است و رابدومیوبلاستهای مخطط دیده میشوند. بیشتر در قسمت تحتانی اربیت رخ میدهد.
پیشآگهی: میزان بقای 5 ساله برای موارد اربیت 95٪ و مطلوب است. میانگین سن شروع 7-8 سال.
نوع آلوئولار
فراوانی: حدود 20٪ از کل رابدومیوسارکومها. حدود 10٪ از رابدومیوسارکومهای اربیت.
تصویر بافتی: تومور به صورت آلوئولار رشد میکند. سلولهای بزرگ نامنظم با هسته بزرگ و سیتوپلاسم فراوان ائوزینوفیل در اطراف ترابکولها قرار میگیرند. تمایل به متاستاز گسترده دارد.
پیشآگهی: میزان بقای 5 ساله برای موارد اربیت 74٪. این تهاجمیترین نوع بافتی با بدترین پیشآگهی است.
نوع پلئومورفیک (تمایز یافته)
فراوانی: در کودکان نادر است. عمدتاً در اندامهای بزرگسالان (به ویژه ران) رخ میدهد.
تصویر بافتی: سلولهای گرد تا کشیده با سیتوپلاسم فراوان و چند هستهای که در آنها مخطط بودن قابل تشخیص است. از سلولهای توموری با پلئومورفیسم بالا تشکیل شده است. فراوانی کم اما درجه تمایز بالا.
پیشآگهی: در موارد اربیت نسبتاً مطلوب است.
نوع خوشهای (botryoid) یک نوع جنینی است که بیشتر در نوزادان دیده میشود و با تجمع سلولهای توموری زیر اپیتلیال مشخص میشود که ظاهری «خوشهای» ایجاد میکند.
محلهای با پیشآگهی خوب: اربیت، دستگاه ادراری تناسلی به جز مثانه و پروستات، سر و گردن به جز پارامننژ
محلهای با پیشآگهی بد: اندامها، مثانه، پروستات، پارامننژ
از نظر بافتشناسی، پیشآگهی نوع آلوئولار و پلئومورفیک نسبت به نوع جنینی بدتر است. میزان بقای ۵ ساله برای کل رابدومیوسارکوم ۶۶٪ است، اما رابدومیوسارکوم اربیت به دلیل محل بروز در گروه پیشآگهی خوب طبقهبندی میشود.
رابدومیوسارکوم آلوئولار با جابهجاییهای کروموزومی مکرر t(2;13)(q35;q14) و t(1;13)(p36;q14) مشخص میشود. حدود ۸۰٪ از رابدومیوسارکومهای آلوئولار دارای جابهجایی PAX3-FOXO1 یا PAX7-FOXO1 هستند. همجوشی PAX3-FOXO1 با بیان بالای OLIG2 و پیشآگهی ضعیف مرتبط است2).
رابدومیوسارکوم جنینی بازآرایی ساختاری کروموزومی مکرر ندارد، اما از دست رفتن آللی در کروموزوم ۱۱ (به ویژه ناحیه 11p15.5) با فراوانی بالا مشاهده میشود.
Qچه تفاوتی در پیشآگهی بین نوع جنینی و آلوئولار وجود دارد؟
A
میزان بقای ۵ ساله برای نوع جنینی رابدومیوسارکوم اربیت ۹۵٪ است، در حالی که برای نوع آلوئولار ۷۴٪ و پایینتر است. نوع آلوئولار با جابهجاییهای کروموزومی مانند PAX3-FOXO1 همراه است و نوع بافتی مستعد متاستاز گسترده میباشد.
۷. تحقیقات جدید و چشماندازهای آینده (گزارشهای در مرحله تحقیق)
در سالهای اخیر، یک زیرگروه جدید به نام رابدومیوسارکوم سلول دوکی اپیتلیوئید با بازآرایی TFCP2 شناسایی شده است.
لی و همکاران (۲۰۲۳) گزارش کردند که رابدومیوسارکوم سلول دوکی اپیتلیوئید عمدتاً در استخوانها (سر و گردن، لگن) رخ میدهد و با همجوشی EWSR1-TFCP2 یا FUS-TFCP2 مشخص میشود که یک زیرگروه با پیشآگهی بسیار بد است3). میانگین بقای موارد گزارش شده تنها ۱۷ ماه است.
تحقیقات درمانی با هدف قرار دادن پروتئین همجوشی PAX3-FOXO1 که در حدود ۸۰٪ از رابدومیوسارکومهای آلوئولار نقش دارد، در حال پیشرفت است.
گزارش شده است که فرمولاسیون حاوی RNA مداخلهگر کوچک (siRNA) درون ذرات لیپوزوم-پروتامین، بیان PAX3-FOXO1 را در ردههای سلولی رابدومیوسارکوم آلوئولار در شرایط آزمایشگاهی به طور مؤثر کاهش میدهد و باعث تأخیر و مهار رشد تومورهای پیوندی رابدومیوسارکوم آلوئولار میشود1).
استفاده از مهارکنندههای ایست بازرسی ایمنی (مانند نیولوماب) برای سارکوم متاستاتیک در حال بررسی است1). با این حال، در حال حاضر اثربخشی آن برای رابدومیوسارکوم تأیید نشده است.
در یک مرور متون بر روی رابدومیوسارکوم آلوئولار اولیه ناحیه غده صنوبری در بزرگسالان، ارتباط بین شدت درمان و طول عمر بقا نشان داده شده است.
در مرور Chang و همکاران (2025) بر روی 13 مورد رابدومیوسارکوم آلوئولار اولیه ناحیه غده صنوبری در بزرگسالان، میانگین بقا با جراحی به تنهایی حدود 5 ماه، با جراحی به همراه پرتودرمانی حدود 10.28 ماه، و با جراحی به همراه پرتودرمانی و شیمیدرمانی حدود 11.33 ماه گزارش شد که نشاندهنده افزایش بقا با درمان ترکیبی است2).
Yang N, Kong D, Wang X, Liu Y. Perianal rhabdomyosarcoma in an adult: A case report and review of the literature. Medicine. 2023;102(48):e36199.
Chang T, Ding C, Liu Y, Yang Y, Mao Q.. Primary pineal alveolar rhabdomyosarcoma in an adult patient: a case report and literature review. BMC Neurol. 2025;25(1):106. doi:10.1186/s12883-025-04113-8. PMID:40082819; PMCID:PMC11905731.
Li Y, Li D, Wang J, Tang J.. Epithelioid and spindle rhabdomyosarcoma with TFCP2 rearrangement in abdominal wall: a distinctive entity with poor prognosis. Diagn Pathol. 2023;18(1):41. doi:10.1186/s13000-023-01330-y. PMID:36998041; PMCID:PMC10061849.
متن مقاله را کپی کنید و در دستیار هوش مصنوعی دلخواه خود بچسبانید.
مقاله در کلیپبورد کپی شد
یکی از دستیارهای هوش مصنوعی زیر را باز کنید و متن کپیشده را در کادر گفتگو بچسبانید.