Rhabdomyosarcoma adalah tumor ganas yang berasal dari sel mesenkim orbita, menunjukkan diferensiasi ke arah otot lurik. Tumor ini timbul dari sel mesenkim yang tidak berdiferensiasi di dalam orbita, bukan dari jaringan otot yang telah berdiferensiasi seperti otot ekstraokular. Ini adalah tumor ganas orbita yang paling sering pada anak-anak, dan harus menjadi diagnosis pertama yang dipertimbangkan ketika melihat tumor orbita yang membesar dengan cepat pada anak.
Di Amerika Serikat, ini mencakup sekitar 5% dari semua kanker anak, dengan sekitar 250 kasus baru didiagnosis setiap tahun. Sekitar 10% di antaranya (sekitar 35 kasus per tahun) berasal dari orbita. Insiden tahunan diperkirakan 4 kasus per juta populasi umum, dan 4,5 kasus per juta anak1).
Usia rata-rata saat diagnosis adalah 7-8 tahun, dan dua pertiga terjadi pada anak di bawah 10 tahun. 90% terjadi sebelum usia 16 tahun, dan sedikit lebih sering pada anak laki-laki (rasio 5:3). Tidak ada perbedaan insiden berdasarkan ras. Tumor ini berbatas tegas dan sering terjadi di orbita bagian atas.
Distribusi lokasi rhabdomyosarcoma okular adalah: orbita 76%, konjungtiva 12%, uvea 9%, dan kelopak mata 3%. Dapat timbul di dalam orbita atau menginvasi orbita dari jaringan sekitarnya seperti sinus paranasal. Orbita merupakan lokasi primer pada 10% dari seluruh rhabdomyosarcoma; lokasi lain termasuk traktus urogenital (22%), ekstremitas (18%), daerah parameningeal (16%), dan kepala dan leher lainnya (10%).
QPada usia berapa rhabdomyosarcoma paling sering terjadi?
A
2/3 kasus terjadi pada anak di bawah 10 tahun, dengan usia rata-rata onset 7-8 tahun. 90% terjadi sebelum usia 16 tahun. Jarang terjadi pada orang dewasa tetapi telah dilaporkan.
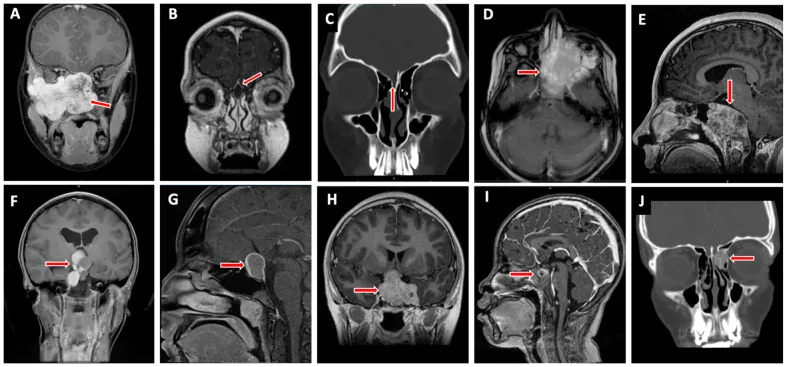
Valencia-Sanchez BA, et al. Special Considerations in Pediatric Endoscopic Skull Base Surgery. J Clin Med. 2024. Figure 2. PMCID: PMC11013018. License: CC BY.
Panah merah menunjukkan rhabdomyosarcoma (D), salah satu lesi khas pada dasar tengkorak anak. Sesuai dengan infiltrasi tumor yang dibahas di bagian “2. Gejala utama dan temuan klinis”.
Proptosis cepat: Proptosis unilateral yang berkembang pesat dalam beberapa minggu adalah gejala khas.
Pembengkakan kelopak mata: Kelopak mata membengkak seiring pertumbuhan tumor. Meskipun pembengkakan parah, kemerahan ringan dan tanda inflamasi sedikit, nyeri tidak hebat, yang merupakan ciri khas.
Keterbatasan gerakan mata, ptosis, dan berbagai gambaran klinis lainnya tergantung lokasi tumor.
Sering terjadi pada anak usia 0-9 tahun, dan ditandai dengan perkembangan yang cepat.
Lesi paling sering terletak di bagian atas atau atas-dalam, dan massa mendorong bola mata ke bawah atau ke luar. Massa berbentuk bulat hingga oval dengan batas yang relatif jelas terlihat di luar kerucut otot.
Sekitar 10% kasus berasal dari sinus paranasal atau rongga hidung dan menginvasi orbita secara sekunder, dapat disertai sinusitis, hidung tersumbat, dan perdarahan hidung. Tumor posterior dapat disertai lipatan koroid, ablasi retina, dan edema papilsaraf optik.
Pada kasus primer konjungtiva, muncul sebagai massa granular berwarna merah muda seperti buah anggur di forniks konjungtiva. Pada kasus primer uvea, muncul sebagai massa iris dan dapat disertai penyemaian bilik anterior atau glaukoma sekunder.
Metastasis paling sering ke paru-paru, diikuti sumsum tulang, tulang, dan kelenjar getah bening. Orbita hampir tidak memiliki pembuluh limfe, tetapi tumor anterior di konjungtiva dan kelopak mata dapat bermetastasis ke kelenjar getah bening regional.
QPenyakit apa saja yang sering disalahartikan sebagai rhabdomyosarcoma?
A
Diagnosis banding klinis meliputi: neuroblastoma, kloroma, limfangioma, hemangioma infantil, selulitis orbita, dan inflamasi nonspesifik. Selain itu, perluasan sinusitis ke orbita, ruptur kista dermoid, sarkoma Ewing, dan infiltrasi leukemia ke orbita juga termasuk dalam diagnosis banding. Diferensiasi dilakukan berdasarkan progresi cepat serta temuan pencitraan dan biopsi.
Rhabdomyosarcoma berasal dari sel mesenkim yang belum berdiferensiasi dengan potensi multipel di jaringan lunak orbita, bukan dari otot ekstraokular. Sebagian besar bersifat sporadis dan penyebab pastinya tidak diketahui.
Faktor risiko genetik yang terkait adalah sebagai berikut:
CT kepala dan orbita dilakukan segera untuk memeriksa adanya tumor di orbita, homogenitas tumor, destruksi dinding tulang orbita, serta perluasan ke intrakranial atau sinus paranasal. Selanjutnya, MRI dilakukan untuk menilai karakteristik internal tumor dan hubungannya dengan bola mata serta otot ekstraokular.
Temuan khas untuk setiap modalitas adalah sebagai berikut:
Pemeriksaan
Temuan Utama
CT
Massa bulat atau oval yang homogen dan berbatas tegas. Efek kontras sedang hingga tinggi
MRI T1
Sinyal rendah terhadap lemak orbita, sinyal sama terhadap otot ekstraokular
Pada MRI, kadang sulit membedakan dengan hemangioma kapiler, tetapi hemangioma kapiler kaya akan pembuluh darah (aliran darah) sehingga dapat dibedakan. Jika komponen stroma banyak, intensitas sinyal MRI berbeda.
Untuk mencari metastasis, dilakukan foto toraks, skintigrafi tulang, dan sitologi aspirasi sumsum tulang. Untuk pencarian sistemik, digunakan PET/CT, CT seluruh tubuh, dan skintigrafi.
Untuk diagnosis pasti diperlukan biopsi. Biopsi aspirasi jarum halus tidak mencukupi, dilakukan biopsi eksisi atau biopsi insisi.
Biopsi eksisi: Dipilih jika tumor dapat diangkat secara bedah tanpa merusak struktur penting.
Biopsi insisi: Dipilih jika tumor besar dan terletak di orbita posterior.
Pada diagnosis cepat intraoperatif, kadang sulit membedakan dari tumor lain. Prinsipnya adalah melakukan biopsi pada operasi darurat dan memulai kemoterapi serta radioterapi sedini mungkin.
Diagnosis ditegakkan dengan pembuktian rhabdomyoblast melalui mikroskop cahaya, imunohistokimia, dan mikroskop elektron.
Garis-garis lurik dideteksi dengan pewarnaan HE dan Masson trichrome. Untuk membedakan sel yang tidak berdiferensiasi, dilakukan pewarnaan imunohistokimia, dan pola desmin positif, HHF-35 (aktin) positif, dan α-smooth muscle actin negatif berguna untuk diagnosis. Mikroskop elektron menunjukkan berkas aktin-miosin, protein pita A, I, dan Z.
Penyakit banding klinis: neuroblastoma, kloroma, limfangioma, hemangioma infantil, selulitis orbita, inflamasi nonspesifik. Selain itu, perluasan sinusitis ke orbita, selulitis orbita, ruptur kista dermoid, perdarahan intratumor hemangioma, perdarahan limfangioma, sarkoma Ewing, infiltrasi leukemia ke orbita (kloroma), dan perdarahan orbita juga termasuk dalam diagnosis banding.
Eksisi total melalui pembedahan tidak mungkin dilakukan dan tidak boleh menjadi tujuan. Saat ini, tujuan utama pembedahan adalah untuk mendapatkan diagnosis jaringan, dan era eksenterasiorbita sebagai pilihan pertama telah berakhir. Setelah diagnosis patologis dikonfirmasi dengan biopsi eksisi, dilakukan CT toraks-abdomen dan biopsi sumsum tulang di bagian pediatri untuk memeriksa adanya metastasis paru atau metastasis sistemik lainnya. Kemoterapi sistemik adalah dasar pengobatan, dan terapi kombinasi tiga modalitas (operasi, radioterapi, dan kemoterapi) adalah terapi standar.
Kemoterapi terutama menggunakan regimen VAC (vinkristin + aktinomisin D + siklofosfamid). Selain itu, manfaat ifosfamid dan etoposid juga telah dilaporkan.
Radioterapi diberikan dengan dosis sekitar 40-60 Gy. Mulai April 2016, terapi proton telah dicakup oleh asuransi kesehatan dan ditambahkan sebagai pilihan terapi standar. Terapi proton memiliki keuntungan mengurangi dosis pada jaringan normal. Transplantasi sel punca hematopoietik juga dilakukan pada beberapa kasus.
Rencana pengobatan untuk rhabdomyosarcoma orbita ditentukan berdasarkan klasifikasi Intergroup Rhabdomyosarcoma Study (IRS). Dalam 30 kasus oleh Shields dkk., distribusi kelompok adalah: Grup I 7%, Grup II 37%, Grup III 53%, Grup IV 3%.
Grup
Definisi
Rencana Pengobatan
I
Eksisi lengkap penyakit lokal
Kemoterapi saja
II
Penyakit sisa atau metastasis kelenjar getah bening regional
Kemoterapi + Radioterapi
III
Reseksi tidak lengkap atau sisa makroskopis
Kemoterapi + Radioterapi
IV
Metastasis jauh
Kemoterapi + Radioterapi
Radioterapi untuk kelompok II hingga IV adalah 4000-5000 cGy (40-50 Gy) selama 4-5 minggu.
Pada kasus yang tidak dapat direseksi total atau kasus rekuren, pertimbangkan radioterapi atau kemoterapi sebagai terapi paliatif. Tindakan bedah dapat berupa eksisi tumor atau eviserasiorbita. Meskipun reseksi total berhasil, kemoterapi tetap wajib. Orbita adalah lokasi dengan prognosis baik; dengan terapi yang tepat, kelangsungan hidup lebih dari 90% dapat diharapkan.
Jika terdapat invasi intrakranial atau sinus paranasal, atau metastasis ke paru-paru atau kelenjar getah bening leher, prognosis kehidupan buruk. Deteksi dini dan inisiasi kemoterapi dini memberikan respons baik terhadap kemoterapi dan prognosis kehidupan yang baik.
Komplikasi oftalmologis dari kemoterapi yang dilaporkan meliputi keratokonjungtivitis sicca dan blefarokonjungtivitis akibat siklofosfamid, konjungtivitis dan penglihatan kabur akibat ifosfamid, serta oklusi arteri retina sentral akibat etoposid.
Kekambuhan lokal dievaluasi dengan melakukan MRI secara teratur. Metastasis hematogen sering terjadi, sehingga dilakukan pemeriksaan sistemik secara teratur.
Hasil penglihatan jangka panjang setelah pengobatan (Shields) adalah: 20/20 hingga 20/40 pada 39%, 20/50 hingga 20/100 pada 18%, dan 20/200 hingga tidak ada persepsi cahaya pada 43%.
QKomplikasi mata apa yang dapat terjadi setelah pengobatan rhabdomyosarcoma?
Rhabdomyosarcoma diklasifikasikan menurut WHO menjadi 4 subtipe: embrional, alveolar, pleomorfik, dan sel spindel/sklerosing 2).
Tipe embrional
Frekuensi: Mencakup 50-60% dari seluruh rhabdomyosarcoma, dan 80-84% dari rhabdomyosarcoma orbita.
Histologi: Berkas sel berbentuk gelendong (sel seperti vili) berjalan ke berbagai arah. Sel berbentuk bulat kecil atau gelendong, dengan pleomorfisme inti yang sedikit. Sitoplasma sedikit dengan granula eosinofilik, dan terdapat rhabdomyoblast dengan lurik. Lebih sering di bagian bawah orbita.
Prognosis: Tingkat kelangsungan hidup 5 tahun untuk kasus orbita adalah 95%, baik. Usia rata-rata onset 7-8 tahun.
Tipe Alveolar
Frekuensi: Sekitar 20% dari seluruh rhabdomyosarcoma. Sekitar 10% dari rhabdomyosarcoma orbita.
Histologi: Tumor tumbuh dalam pola alveolar. Sel besar tidak beraturan dengan inti besar dan sitoplasma kaya yang terwarnai eosin, tersusun di sekitar trabekula. Rentan terhadap metastasis luas.
Prognosis: Tingkat kelangsungan hidup 5 tahun untuk kasus orbita adalah 74%. Ini adalah tipe histologis dengan keganasan tertinggi dan prognosis terburuk.
Tipe Pleomorfik (Berdiferensiasi)
Frekuensi: Jarang pada anak-anak. Terutama terjadi pada ekstremitas (khususnya paha) orang dewasa.
Histologi: Sel bulat hingga memanjang dengan inti banyak dan sitoplasma kaya, lurik dapat diidentifikasi. Terdiri dari sel tumor yang sangat pleomorfik. Frekuensi rendah tetapi derajat diferensiasi tinggi.
Prognosis: Relatif baik pada kasus orbita.
Tipe Botryoid adalah varian dari tipe embrional yang sering terjadi pada bayi, ditandai dengan agregat sel tumor subepitel yang memberikan penampilan seperti “anggur”.
Lokasi prognosis baik: Orbita, traktus urogenitalis kecuali kandung kemih dan prostat, kepala dan leher kecuali parameningeal
Lokasi prognosis buruk: Ekstremitas, kandung kemih, prostat, parameningeal
Secara histologis, prognosis tipe alveolar dan pleomorfik lebih buruk dibandingkan tipe embrional. Tingkat kelangsungan hidup 5 tahun untuk rhabdomyosarcoma secara keseluruhan adalah 66%, namun rhabdomyosarcoma orbita diklasifikasikan ke dalam kelompok prognosis baik berdasarkan lokasi terjadinya.
Rhabdomyosarcoma alveolar ditandai dengan translokasi kromosom berulang t(2;13)(q35;q14) dan t(1;13)(p36;q14). Sekitar 80% rhabdomyosarcoma alveolar melibatkan translokasi PAX3-FOXO1 atau PAX7-FOXO1. Fusi PAX3-FOXO1 terkait dengan ekspresi tinggi OLIG2 dan prognosis buruk2).
Rhabdomyosarcoma embrional tidak memiliki penataan ulang struktural kromosom yang berulang, namun kehilangan alel pada kromosom 11 (terutama wilayah 11p15.5) sering diamati.
QApa perbedaan prognosis antara tipe embrional dan alveolar?
A
Tingkat kelangsungan hidup 5 tahun untuk tipe embrional pada rhabdomyosarcoma orbita adalah 95%, sedangkan tipe alveolar adalah 74% lebih rendah. Tipe alveolar disertai translokasi kromosom seperti PAX3-FOXO1 dan merupakan tipe histologis yang cenderung bermetastasis luas.
7. Penelitian terbaru dan prospek masa depan (laporan tahap penelitian)
Dalam beberapa tahun terakhir, subtipe baru yang disebut rhabdomyosarcoma sel spindel epiteloid dengan rearransemen TFCP2 telah diidentifikasi.
Li dkk. (2023) melaporkan bahwa rhabdomyosarcoma sel spindel epiteloid sering terjadi di tulang (kepala-leher, panggul) dan ditandai dengan fusi EWSR1-TFCP2 atau FUS-TFCP2, subtipe dengan prognosis sangat buruk3). Median kelangsungan hidup kasus yang dilaporkan hanya 17 bulan.
Terapi target molekuler yang menargetkan PAX3-FOXO1
Penelitian terapi yang menargetkan protein fusi PAX3-FOXO1, yang terlibat dalam sekitar 80% rhabdomyosarcoma alveolar, sedang berlangsung.
Telah dilaporkan bahwa formulasi yang mengandung small interfering RNA (siRNA) dalam partikel liposom-protamin secara efisien menurunkan regulasi ekspresi PAX3-FOXO1 pada sel rhabdomyosarcoma alveolar in vitro, dan menyebabkan keterlambatan atau penekanan pertumbuhan tumor xenograft rhabdomyosarcoma alveolar1).
Penghambat checkpoint imun (seperti nivolumab) sedang dicoba untuk sarkoma metastatik1). Namun, efektivitasnya pada rhabdomyosarcoma belum terbukti saat ini.
Dalam tinjauan literatur tentang rhabdomyosarcoma alveolar primer di daerah kelenjar pineal pada orang dewasa, hubungan antara intensitas pengobatan dan kelangsungan hidup telah disarankan.
Dalam tinjauan Chang dkk. (2025) terhadap 13 kasus rhabdomyosarcoma alveolar primer kelenjar pineal pada orang dewasa, dilaporkan bahwa rata-rata kelangsungan hidup dengan operasi saja sekitar 5 bulan, dengan operasi + radioterapi sekitar 10,28 bulan, dan dengan operasi + radioterapi + kemoterapi sekitar 11,33 bulan, menunjukkan bahwa pengobatan kombinasi memperpanjang kelangsungan hidup2).
Yang N, Kong D, Wang X, Liu Y. Perianal rhabdomyosarcoma in an adult: A case report and review of the literature. Medicine. 2023;102(48):e36199.
Chang T, Ding C, Liu Y, Yang Y, Mao Q.. Primary pineal alveolar rhabdomyosarcoma in an adult patient: a case report and literature review. BMC Neurol. 2025;25(1):106. doi:10.1186/s12883-025-04113-8. PMID:40082819; PMCID:PMC11905731.
Li Y, Li D, Wang J, Tang J.. Epithelioid and spindle rhabdomyosarcoma with TFCP2 rearrangement in abdominal wall: a distinctive entity with poor prognosis. Diagn Pathol. 2023;18(1):41. doi:10.1186/s13000-023-01330-y. PMID:36998041; PMCID:PMC10061849.
Salin teks artikel dan tempelkan ke asisten AI pilihan Anda.
Artikel disalin ke papan klip
Buka asisten AI di bawah, lalu tempelkan teks yang disalin ke kotak chat.