Rhabdomyosarcoma (RMS ) เป็นเนื้องอกร้ายในเบ้าตา ที่พบบ่อยที่สุดในเด็ก
ตาโปนข้างเดียวที่ดำเนินไปอย่างรวดเร็วภายในไม่กี่สัปดาห์เป็นอาการทั่วไป
อายุที่พบบ่อยคือ 7-8 ปี และ 90% เกิดขึ้นก่อนอายุ 16 ปี
การวินิจฉัยที่แน่นอนต้องใช้การตัดชิ้นเนื้อ การเจาะดูดด้วยเข็มเล็กไม่เพียงพอ
การรักษามาตรฐานคือการผ่าตัดร่วมกับการฉายรังสีและเคมีบำบัด (เช่น สูตร VAC)
การรักษาด้วยโปรตอนเริ่มครอบคลุมโดยประกันตั้งแต่เดือนเมษายน 2016 และถูกเพิ่มเข้าไปในทางเลือกการรักษามาตรฐาน
การพยากรณ์โรคแตกต่างกันตามชนิดเนื้อเยื่อ ชนิดตัวอ่อนในเบ้าตา มีรายงานอัตราการรอดชีวิต 5 ปี 95%
การจัดการภาวะแทรกซ้อนระยะยาว เช่น ต้อกระจก และจอประสาทตา เสื่อมหลังการฉายรังสีเป็นสิ่งสำคัญ
Rhabdomyosarcoma เป็นเนื้องอกร้ายที่เกิดจากเซลล์มีเซนไคม์ในเบ้าตา แสดงการเปลี่ยนแปลงไปทางกล้ามเนื้อลาย เกิดจากเซลล์มีเซนไคม์ที่ไม่มีการเปลี่ยนแปลงในเบ้าตา ไม่ใช่จากเนื้อเยื่อกล้ามเนื้อที่เปลี่ยนแปลงแล้ว เช่น กล้ามเนื้อนอกลูกตา เป็นเนื้องอกร้ายในเบ้าตา ที่พบบ่อยที่สุดในเด็ก และควรเป็นโรคแรกที่คิดถึงเมื่อพบเนื้องอกในเบ้าตา ที่ขยายตัวอย่างรวดเร็วในเด็ก
ในสหรัฐอเมริกา คิดเป็นประมาณ 5% ของมะเร็งเด็กทั้งหมด โดยมีผู้ป่วยรายใหม่ประมาณ 250 รายต่อปี ประมาณ 10% (ประมาณ 35 รายต่อปี) เป็นมะเร็งปฐมภูมิในเบ้าตา อุบัติการณ์รายปีประมาณ 4 รายต่อประชากรล้านคน และ 4.5 รายต่อเด็กล้านคน1)
อายุเฉลี่ยเมื่อวินิจฉัยคือ 7-8 ปี และสองในสามเกิดขึ้นในเด็กอายุต่ำกว่า 10 ปี 90% เกิดขึ้นก่อนอายุ 16 ปี และพบในเด็กผู้ชายมากกว่าเล็กน้อย (อัตราส่วน 5:3) ไม่มีความแตกต่างของอุบัติการณ์ตามเชื้อชาติ เนื้องอกมีขอบเขตชัดเจนและมักเกิดในส่วนบนของเบ้าตา
การกระจายตำแหน่งของ rhabdomyosarcoma ในตา: เบ้าตา 76%, เยื่อบุตา 12%, ยูเวีย 9%, เปลือกตา 3% อาจเกิดขึ้นภายในเบ้าตา หรือลุกลามเข้าสู่เบ้าตา จากเนื้อเยื่อรอบข้าง เช่น โพรงอากาศข้างจมูก เบ้าตา เป็นตำแหน่งปฐมภูมิของ rhabdomyosarcoma ทั้งหมด 10% ตำแหน่งอื่นๆ ได้แก่ ทางเดินปัสสาวะและอวัยวะสืบพันธุ์ (22%), แขนขา (18%), บริเวณใกล้เยื่อหุ้มสมอง (16%), และศีรษะและคออื่นๆ (10%)
Q
Rhabdomyosarcoma เกิดขึ้นบ่อยที่สุดในช่วงอายุใด?
A
2 ใน 3 ของผู้ป่วยเกิดในเด็กอายุต่ำกว่า 10 ปี อายุเฉลี่ยที่เริ่มป่วยคือ 7-8 ปี 90% เกิดก่อนอายุ 16 ปี พบได้น้อยในผู้ใหญ่แต่มีรายงาน
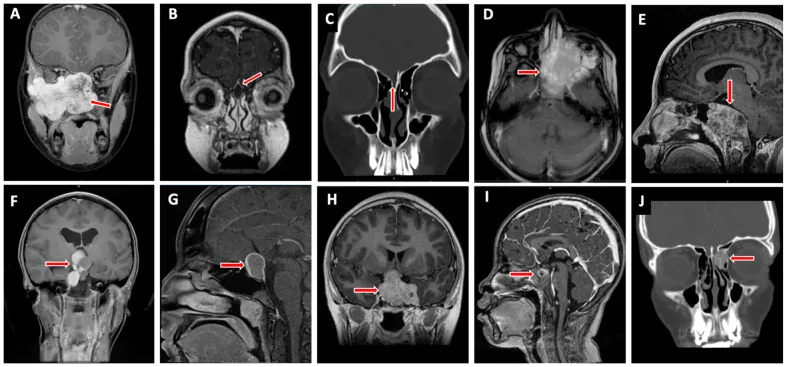
Valencia-Sanchez BA, et al. Special Considerations in Pediatric Endoscopic Skull Base Surgery. J Clin Med. 2024. Figure 2. PM
CI D: PMC11013018. License: CC BY.
ลูกศรสีแดงชี้ไปที่ rhabdomyosarcoma (D) ซึ่งเป็นหนึ่งในรอยโรคทั่วไปที่ฐานกะโหลกศีรษะในเด็ก สอดคล้องกับการแทรกซึมของเนื้องอกที่กล่าวถึงในหัวข้อ “2. อาการหลักและอาการแสดงทางคลินิก”
ตาโปนเร็ว : ตาโปนข้างเดียวที่ดำเนินไปอย่างรวดเร็วภายในไม่กี่สัปดาห์เป็นอาการทั่วไปเปลือกตาบวม : เปลือกตาบวมเมื่อเนื้องอกโตขึ้น แม้บวมมาก แต่แดงเล็กน้อยและมีอาการอักเสบน้อย ปวดไม่รุนแรง ซึ่งเป็นลักษณะเฉพาะการจำกัดการเคลื่อนไหวของลูกตา หนังตาตก และลักษณะทางคลินิกอื่นๆ ขึ้นอยู่กับตำแหน่งของเนื้องอก
พบบ่อยในเด็กอายุ 0-9 ปี และมีลักษณะการดำเนินโรคที่รวดเร็ว
ความถี่ของอาการแสดงตามรายงานของ Shields และคณะมีดังนี้:
อาการแสดง ความถี่ ตาโปน 80-100% ตาเบน 80% เยื่อบุตา บวมและเปลือกตาบวม60% หนังตาตก 30-50% ก้อนที่คลำได้ 25% ปวด 10%
รอยโรคมักอยู่ด้านบนหรือด้านบน-ด้านใน ก้อนเนื้องอกจะเบนลูกตาไปด้านล่างหรือด้านนอก พบก้อนกลมถึงรูปไข่ ขอบเขตค่อนข้างชัดเจน อยู่นอกกรวยกล้ามเนื้อ
ประมาณ 10% ของกรณีเกิดจากไซนัสพารานาซัลหรือโพรงจมูกและบุกรุกเบ้าตา แบบทุติยภูมิ อาจมีไซนัสอักเสบ คัดจมูก เลือดกำเดาไหล เนื้องอกด้านหลังอาจมีรอยพับของคอรอยด์ จอประสาทตาลอก และจานประสาทตา บวมน้ำ
กรณีปฐมภูมิที่เยื่อบุตา จะปรากฏเป็นก้อนเนื้อเม็ดสีชมพูคล้ายพวงองุ่นที่รอยต่อเยื่อบุตา กรณีปฐมภูมิที่ยูเวีย จะปรากฏเป็นก้อนที่ม่านตา และอาจมีการกระจายในช่องหน้าหรือต้อหินทุติยภูมิ
การแพร่กระจายที่พบบ่อยที่สุดคือไปยังปอด รองลงมาคือไขกระดูก กระดูก และต่อมน้ำเหลือง เบ้าตา แทบไม่มีท่อน้ำเหลือง แต่เนื้องอกด้านหน้าที่เยื่อบุตา และเปลือกตาสามารถแพร่กระจายไปยังต่อมน้ำเหลืองเฉพาะที่ได้
Q
โรคใดบ้างที่มักสับสนกับ rhabdomyosarcoma?
A
การวินิจฉัยแยกโรคทางคลินิก ได้แก่ นิวโรบลาสโตมา คลอโรมา ลิมแฟงจิโอมา ฮีแมงจิโอมาในทารก เซลลูไลติสของเบ้าตา และการอักเสบที่ไม่จำเพาะ นอกจากนี้ การลุกลามของไซนัสอักเสบเข้าสู่เบ้าตา การแตกของถุงน้ำเดอร์มอยด์ ยูวิงซาร์โคมา และการแทรกซึมของมะเร็งเม็ดเลือดขาวเข้าสู่เบ้าตา ก็รวมอยู่ในการวินิจฉัยแยกโรคด้วย การแยกโรคอาศัยการดำเนินโรคที่รวดเร็วและผลการตรวจภาพและการตัดชิ้นเนื้อ
แรบโดไมโอซาร์โคมาเกิดจากเซลล์มีเซนไคม์ที่ยังไม่แยกตัวซึ่งมีศักยภาพหลายทางในเนื้อเยื่ออ่อนของเบ้าตา ไม่ใช่จากกล้ามเนื้อนอกลูกตา ส่วนใหญ่เกิดขึ้นเป็นประปรายและไม่ทราบสาเหตุที่แน่ชัด
ปัจจัยเสี่ยงทางพันธุกรรมที่เกี่ยวข้องมีดังนี้:
นิวโรไฟโบรมาโตซิสชนิดที่ 1 : ทราบว่ามีความสัมพันธ์กับความเสี่ยงที่เพิ่มขึ้นของแรบโดไมโอซาร์โคมากลุ่มอาการลิ-ฟราวเมนี : เกี่ยวข้องกับความผิดปกติของยีนยับยั้งเนื้องอก p53กลุ่มอาการเบ็ควิท-วีเดอมันน์ : โรคแต่กำเนิดที่มีลักษณะการเจริญเติบโตเกิน ไส้เลื่อนสะดือ และน้ำตาลในเลือดต่ำรีติโนบลาสโตมา ทางพันธุกรรม
ทำซีทีสแกนศีรษะและเบ้าตา อย่างเร่งด่วนเพื่อตรวจสอบว่ามีเนื้องอกในเบ้าตา หรือไม่ ความสม่ำเสมอของเนื้องอก การทำลายผนังกระดูกเบ้าตา และการลุกลามเข้าไปในกะโหลกศีรษะหรือไซนัสพารานาซัล จากนั้นทำเอ็มอาร์ไอเพื่อประเมินลักษณะภายในของเนื้องอกและความสัมพันธ์กับลูกตาและกล้ามเนื้อนอกลูกตา
ลักษณะเฉพาะของแต่ละวิธีการตรวจมีดังนี้:
การตรวจ ผลการตรวจหลัก ซีทีสแกน ก้อนเนื้อกลมหรือรี เนื้อแน่น ขอบเขตชัดเจน มีการเพิ่มความเข้มของสารทึบรังสีระดับปานกลางถึงสูง MRI T1 สัญญาณต่ำเมื่อเทียบกับไขมันในเบ้าตา สัญญาณเท่ากับกล้ามเนื้อนอกลูกตา MRI T2 สัญญาณสูงเมื่อเทียบกับไขมันในเบ้าตา และกล้ามเนื้อนอกลูกตา
ในการตรวจ MRI บางครั้งอาจแยกจาก hemangioma ชนิด capillary ได้ยาก แต่ hemangioma ชนิด capillary มีหลอดเลือด (การไหลเวียนเลือด) มาก จึงสามารถแยกได้ หากมีส่วนประกอบของเนื้อเยื่อคั่นระหว่างหนาแน่น ความเข้มสัญญาณ MRI จะแตกต่างกัน
เพื่อค้นหาการแพร่กระจาย จะทำการเอกซเรย์ทรวงอก การสแกนกระดูก และการตรวจเซลล์จากการเจาะไขกระดูก สำหรับการค้นหาทั่วร่างกาย จะใช้ PET/CT, CT ทั่วร่างกาย และการสแกนนิวเคลียร์
เพื่อการวินิจฉัยที่แน่นอน จำเป็นต้องตัดชิ้นเนื้อตรวจ การเจาะดูดด้วยเข็มเล็กไม่เพียงพอ จะทำการตัดชิ้นเนื้อแบบ excision หรือ incision
การตัดชิ้นเนื้อแบบ excision : เลือกเมื่อสามารถผ่าตัดเอาก้อนเนื้อออกได้โดยไม่ทำลายโครงสร้างสำคัญการตัดชิ้นเนื้อแบบ incision : เลือกเมื่อก้อนเนื้อมีขนาดใหญ่และอยู่บริเวณเบ้าตา ส่วนหลัง
ในการวินิจฉัยระหว่างผ่าตัดแบบรวดเร็ว บางครั้งอาจแยกจากเนื้องอกอื่นได้ยาก หลักการคือทำการตัดชิ้นเนื้อในการผ่าตัดฉุกเฉินและเริ่มให้เคมีบำบัดและรังสีรักษาโดยเร็ว
การวินิจฉัยขึ้นอยู่กับการพิสูจน์เซลล์ rhabdomyoblast ด้วยกล้องจุลทรรศน์แบบใช้แสง การย้อมอิมมูโนฮิสโตเคมี และกล้องจุลทรรศน์อิเล็กตรอน
ตรวจพบลายกล้ามเนื้อลายด้วยการย้อม HE และ Masson trichrome เพื่อแยกความแตกต่างของเซลล์ที่ไม่มีการแบ่งตัว จะทำการย้อมอิมมูโนฮิสโตเคมี และรูปแบบที่เดสมีนบวก HHF-35 (แอคติน) บวก และ α-smooth muscle actin ลบ มีประโยชน์ในการวินิจฉัย กล้องจุลทรรศน์อิเล็กตรอนเผยให้เห็นมัดแอคติน-ไมโอซิน และโปรตีนของแถบ A, I, Z
โรคที่ต้องแยกทางคลินิก: นิวโรบลาสโตมา , คลอโรมา, ลิมแฟงจิโอมา, ฮีแมงจิโอมาในทารก, เซลลูไลติสของเบ้าตา , การอักเสบที่ไม่จำเพาะ นอกจากนี้ การลุกลามของไซนัสอักเสบเข้าสู่เบ้าตา , เซลลูไลติสของเบ้าตา , การแตกของเดอร์มอยด์ ซีสต์, การตกเลือดในก้อนฮีแมงจิโอมา, การตกเลือดของลิมแฟงจิโอมา, ยูวิงซาร์โคมา, การแทรกซึมของมะเร็งเม็ดเลือดขาวในเบ้าตา (คลอโรมา), และการตกเลือดในเบ้าตา ก็รวมอยู่ในการวินิจฉัยแยกโรคด้วย
การผ่าตัดเอาออกทั้งหมดเป็นไปไม่ได้และไม่ควรเป็นเป้าหมาย ปัจจุบัน วัตถุประสงค์หลักของการผ่าตัดคือเพื่อให้ได้การวินิจฉัยทางเนื้อเยื่อ และยุคที่ถือว่าการตัดเอาสิ่งในเบ้าตา ออกเป็นทางเลือกแรกได้สิ้นสุดลงแล้ว หลังจากยืนยันการวินิจฉัยทางพยาธิวิทยาด้วยการตัดชิ้นเนื้อเพื่อตรวจ จะทำ CT ทรวงอก-ช่องท้อง และเจาะไขกระดูกในแผนกกุมารเวชศาสตร์เพื่อตรวจหาการแพร่กระจายไปยังปอดหรือทั่วร่างกาย การให้เคมีบำบัดทั่วร่างกายเป็นพื้นฐานของการรักษา และการรักษาแบบผสมผสานสามวิธี (การผ่าตัด การฉายรังสี และเคมีบำบัด) เป็นการรักษามาตรฐาน
ในเคมีบำบัด สูตร VAC (วินคริสทีน + แอคติโนมัยซิน D + ไซโคลฟอสฟาไมด์) ถูกใช้เป็นหลัก นอกจากนี้ ยังมีรายงานประโยชน์ของไอฟอสฟาไมด์และอีโทโพไซด์ด้วย
การฉายรังสีให้ในขนาดประมาณ 40-60 เกรย์ ตั้งแต่เดือนเมษายน 2559 การรักษาด้วยโปรตอนได้รับการคุ้มครองโดยประกันสุขภาพและถูกเพิ่มเป็นทางเลือกในการรักษามาตรฐาน การรักษาด้วยโปรตอนมีข้อดีคือลดปริมาณรังสีต่อเนื้อเยื่อปกติ การปลูกถ่ายเซลล์ต้นกำเนิดเม็ดเลือดก็ทำในบางกรณี
แผนการรักษาสำหรับแรบโดไมโอซาร์โคมาของเบ้าตา ถูกกำหนดตามการจำแนกของ Intergroup Rhabdomyosarcoma Study (IRS) ใน 30 รายของ Shields และคณะ การกระจายกลุ่มคือ: กลุ่ม I 7%, กลุ่ม II 37%, กลุ่ม III 53%, กลุ่ม IV 3%
กลุ่ม คำจำกัดความ แผนการรักษา I การตัดออกทั้งหมดของโรคเฉพาะที่ เคมีบำบัดเท่านั้น II โรคหลงเหลือหรือการแพร่กระจายไปยังต่อมน้ำเหลืองในภูมิภาค เคมีบำบัด + รังสีรักษา III การตัดออกไม่สมบูรณ์หรือมีก้อนเหลือที่มองเห็นได้ เคมีบำบัด + รังสีรักษา IV การแพร่กระจายระยะไกล เคมีบำบัด + รังสีรักษา
การฉายรังสีสำหรับกลุ่ม II ถึง IV คือ 4000-5000 cGy (40-50 Gy) ในระยะเวลา 4-5 สัปดาห์
ในกรณีที่ไม่สามารถตัดออกได้ทั้งหมดหรือเป็นซ้ำ ให้พิจารณาการฉายรังสีหรือเคมีบำบัดเป็นการรักษาแบบประคับประคอง การผ่าตัดอาจรวมถึงการตัดก้อนเนื้องอกหรือการเอาสิ่งในเบ้าตา ออก แม้จะตัดออกได้หมดก็ยังต้องให้เคมีบำบัด เบ้าตา เป็นตำแหน่งที่มีพยากรณ์โรคดี ด้วยการรักษาที่เหมาะสม คาดว่าอัตราการรอดชีวิตมากกว่า 90%
หากมีการลุกลามเข้าไปในกะโหลกศีรษะหรือไซนัสพารานาซัล หรือแพร่กระจายไปยังปอดหรือต่อมน้ำเหลืองที่คอ พยากรณ์โรคต่อชีวิตไม่ดี การตรวจพบแต่เนิ่นๆ และเริ่มเคมีบำบัดเร็วจะตอบสนองต่อเคมีบำบัดได้ดีและพยากรณ์โรคต่อชีวิตดี
ภาวะแทรกซ้อนหลักของการฉายรังสี (4000-5000 cGy) มีดังนี้:
ภาวะแทรกซ้อนทางตาจากเคมีบำบัดที่รายงาน ได้แก่ กระจกตา และเยื่อบุตาอักเสบ แห้ง และเปลือกตาและเยื่อบุตาอักเสบ จากไซโคลฟอสฟาไมด์ เยื่อบุตาอักเสบ และตามัวจากไอฟอสฟาไมด์ และหลอดเลือดแดงจอประสาทตา ส่วนกลางอุดตันจากอีโทโพไซด์
การกลับเป็นซ้ำเฉพาะที่ประเมินโดยการตรวจ MRI เป็นระยะ การแพร่กระจายทางกระแสเลือดเกิดขึ้นบ่อย จึงต้องตรวจร่างกายอย่างเป็นระบบเป็นประจำ
ผลลัพธ์การมองเห็น ระยะยาวหลังการรักษา (Shields) คือ: 20/20 ถึง 20/40 ใน 39%, 20/50 ถึง 20/100 ใน 18% และ 20/200 ถึงไม่มีการรับรู้แสงใน 43%
การติดตามทางจักษุวิทยาเป็นประจำสำหรับภาวะแทรกซ้อนระยะยาว เช่น จอประสาทตา เสื่อม ต้อกระจก และตาแห้ง หลังการฉายรังสีเป็นสิ่งจำเป็น หลังสิ้นสุดการรักษา ให้ตรวจตาอย่างครอบคลุมทุก 3-4 เดือนในตอนแรก หลังจากหนึ่งปีให้ตรวจทุก 4-6 เดือน จากนั้นตรวจทุกปีต่อไป
Q
ภาวะแทรกซ้อนทางตาชนิดใดที่อาจเกิดขึ้นหลังการรักษา rhabdomyosarcoma?
Rhabdomyosarcoma จำแนกตาม WHO เป็น 4 ชนิดย่อย: ชนิดเอ็มบริโอนัล ชนิดอัลวีโอลาร์ ชนิดเพลโอมอร์ฟิก และชนิดเซลล์รูปกระสวย/สเกลอโรซิง 2)
ชนิดเอ็มบริโอนัล
ความถี่ : คิดเป็น 50-60% ของ rhabdomyosarcoma ทั้งหมด และ 80-84% ของ rhabdomyosarcoma ในเบ้าตา
ลักษณะเนื้อเยื่อ : มัดของเซลล์รูปกระสวย (เซลล์คล้ายวิลลัส) วิ่งไปในทิศทางต่างๆ เซลล์กลมเล็กหรือรูปกระสวย มีความหลากหลายของนิวเคลียสน้อย ไซโทพลาซึมน้อย มีแกรนูลอีโอซิโนฟิลิก และพบ rhabdomyoblast ที่มีลาย พบบ่อยในส่วนล่างของเบ้าตา
การพยากรณ์โรค : อัตราการรอดชีวิต 5 ปีในกรณีเบ้าตา คือ 95% ซึ่งดี อายุเฉลี่ยที่เริ่มป่วย 7-8 ปี
ชนิดถุงลม (Alveolar)
ความถี่ : ประมาณ 20% ของ rhabdomyosarcoma ทั้งหมด ประมาณ 10% ของ rhabdomyosarcoma ในเบ้าตา
ลักษณะเนื้อเยื่อ : เนื้องอกเจริญแบบถุงลม เซลล์ขนาดใหญ่ไม่สม่ำเสมอมีนิวเคลียสใหญ่และไซโทพลาซึมมากซึ่งติดสีอีโอซิน เรียงตัวรอบ trabeculae มีแนวโน้มแพร่กระจายเป็นวงกว้าง
การพยากรณ์โรค : อัตราการรอดชีวิต 5 ปีในกรณีเบ้าตา คือ 74% นี่เป็นชนิดเนื้อเยื่อที่มีความร้ายแรงสูงสุดและการพยากรณ์โรคแย่ที่สุด
ชนิดหลายรูปร่าง (Pleomorphic) (ชนิดเจริญเต็มที่)
ความถี่ : พบน้อยในเด็ก เกิดที่แขนขาของผู้ใหญ่เป็นหลัก (โดยเฉพาะต้นขา)
ลักษณะเนื้อเยื่อ : เซลล์กลมถึงยาว มีหลายนิวเคลียส ไซโทพลาซึมมาก สามารถระบุลายได้ ประกอบด้วยเซลล์เนื้องอกที่มีความหลากหลายสูง ความถี่ต่ำแต่ระดับการเจริญเต็มที่สูง
การพยากรณ์โรค : ค่อนข้างดีในกรณีเบ้าตา
ชนิดพวงองุ่น (Botryoid) เป็นตัวแปรของชนิดตัวอ่อนที่พบบ่อยในทารก มีลักษณะเฉพาะคือกลุ่มเซลล์เนื้องอกใต้เยื่อบุผิวที่ให้ลักษณะ “คล้ายพวงองุ่น”
ตำแหน่งพยากรณ์โรคดี : เบ้าตา , ทางเดินปัสสาวะและอวัยวะสืบพันธุ์ยกเว้นกระเพาะปัสสาวะและต่อมลูกหมาก, ศีรษะและคอยกเว้นพาราเมนินเจียลตำแหน่งพยากรณ์โรคไม่ดี : แขนขา, กระเพาะปัสสาวะ, ต่อมลูกหมาก, พาราเมนินเจียล
ในทางจุลกายวิภาคศาสตร์ การพยากรณ์โรคของชนิดถุงลมและชนิดหลายรูปร่างแย่กว่าชนิดตัวอ่อน อัตราการรอดชีวิต 5 ปีของแรบโดไมโอซาร์โคมาโดยรวมคือ 66% แต่แรบโดไมโอซาร์โคมาบริเวณเบ้าตา จัดอยู่ในกลุ่มที่มีการพยากรณ์โรคดีตามตำแหน่งที่เกิด
แรบโดไมโอซาร์โคมาชนิดถุงลมมีลักษณะเฉพาะคือการโยกย้ายโครโมโซมซ้ำๆ t(2;13)(q35;q14) และ t(1;13)(p36;q14) ประมาณ 80% ของแรบโดไมโอซาร์โคมาชนิดถุงลมเกี่ยวข้องกับการโยกย้าย PAX3-FOXO1 หรือ PAX7-FOXO1 การหลอมรวม PAX3-FOXO1 สัมพันธ์กับการแสดงออกของ OLIG2 สูงและการพยากรณ์โรคที่ไม่ดี2)
แรบโดไมโอซาร์โคมาชนิดตัวอ่อนไม่มีการจัดเรียงโครงสร้างโครโมโซมซ้ำๆ แต่การสูญเสียอัลลีลบนโครโมโซม 11 (โดยเฉพาะบริเวณ 11p15.5) พบได้บ่อย
Q
การพยากรณ์โรคระหว่างชนิดตัวอ่อนและชนิดถุงลมแตกต่างกันอย่างไร?
A
อัตราการรอดชีวิต 5 ปีของชนิดตัวอ่อนในแรบโดไมโอซาร์โคมาบริเวณเบ้าตา คือ 95% ในขณะที่ชนิดถุงลมคือ 74% ซึ่งต่ำกว่า ชนิดถุงลมมาพร้อมกับการโยกย้ายโครโมโซมเช่น PAX3-FOXO1 และเป็นชนิดเนื้อเยื่อที่มีแนวโน้มแพร่กระจายเป็นวงกว้าง
เนื้อหาต่อไปนี้อยู่ในระยะวิจัยหรือการทดลองทางคลินิกในปัจจุบัน และไม่ใช่การรักษามาตรฐานที่ได้รับในโรงพยาบาลทั่วไป เป็นข้อมูลอ้างอิงสำหรับผู้เชี่ยวชาญเกี่ยวกับความก้าวหน้าทางการแพทย์ในอนาคต
ในช่วงไม่กี่ปีที่ผ่านมา มีการระบุชนิดย่อยใหม่ที่เรียกว่าแรบโดไมโอซาร์โคมาชนิดเซลล์รูปแกนแบบเยื่อบุผิวที่มีการจัดเรียงใหม่ของ TFCP2
Li และคณะ (2023) รายงานว่าแรบโดไมโอซาร์โคมาชนิดเซลล์รูปแกนแบบเยื่อบุผิวมักเกิดที่กระดูก (ศีรษะ-คอ, เชิงกราน) และมีลักษณะเฉพาะคือการหลอมรวม EWSR1-TFCP2 หรือ FUS-TFCP2 ซึ่งเป็นชนิดย่อยที่มีการพยากรณ์โรคแย่มาก3) อายุมัธยฐานการรอดชีวิตของกรณีที่รายงานคือเพียง 17 เดือน
การวิจัยการรักษาที่มุ่งเป้าไปที่โปรตีนหลอมรวม PAX3-FOXO1 ซึ่งเกี่ยวข้องกับประมาณ 80% ของแรบโดไมโอซาร์โคมาชนิดถุงลมกำลังดำเนินอยู่
มีรายงานว่าสูตรยาที่ประกอบด้วย small interfering RNA (siRNA) ที่บรรจุในอนุภาคไลโปโซม-โปรตามีน สามารถลดการแสดงออกของ PAX3-FOXO1 ได้อย่างมีประสิทธิภาพในเซลล์ rhabdomyosarcoma ชนิด alveolar ในหลอดทดลอง และทำให้เกิดการชะลอหรือยับยั้งการเติบโตของเนื้องอก xenograft rhabdomyosarcoma ชนิด alveolar1)
มีการทดลองใช้ยับยั้งจุดตรวจภูมิคุ้มกัน (เช่น nivolumab) ใน sarcoma ที่มีการแพร่กระจาย1) อย่างไรก็ตาม ยังไม่มีการพิสูจน์ประสิทธิภาพใน rhabdomyosarcoma ในขณะนี้
ในการทบทวนวรรณกรรมเกี่ยวกับ rhabdomyosarcoma ชนิด alveolar ปฐมภูมิบริเวณต่อมไพเนียลในผู้ใหญ่ มีการเสนอแนะความสัมพันธ์ระหว่างความเข้มข้นของการรักษาและการรอดชีวิต
ในการทบทวนของ Chang และคณะ (2025) ในผู้ใหญ่ 13 รายที่เป็น rhabdomyosarcoma ชนิด alveolar ปฐมภูมิของต่อมไพเนียล รายงานว่าระยะเวลารอดชีวิตเฉลี่ยด้วยการผ่าตัดเพียงอย่างเดียวประมาณ 5 เดือน ด้วยการผ่าตัดร่วมกับการฉายรังสีประมาณ 10.28 เดือน และด้วยการผ่าตัดร่วมกับการฉายรังสีและเคมีบำบัดประมาณ 11.33 เดือน ซึ่งบ่งชี้ว่าการรักษาแบบผสมผสานช่วยยืดระยะเวลารอดชีวิต2)
Yang N, Kong D, Wang X, Liu Y. Perianal rhabdomyosarcoma in an adult: A case report and review of the literature. Medicine. 2023;102(48):e36199.
Chang T, Ding C, Liu Y, Yang Y, Mao Q.. Primary pineal alveolar rhabdomyosarcoma in an adult patient: a case report and literature review. BMC Neurol. 2025;25(1):106. doi:10.1186/s12883-025-04113-8. PMID:40082819; PMCI D:PMC11905731.
Li Y, Li D, Wang J, Tang J.. Epithelioid and spindle rhabdomyosarcoma with TFCP2 rearrangement in abdominal wall: a distinctive entity with poor prognosis. Diagn Pathol. 2023;18(1):41. doi:10.1186/s13000-023-01330-y. PMID:36998041; PMCI D:PMC10061849.