El rabdomiosarcoma es un tumor maligno derivado de células mesenquimales de la órbita, que muestra diferenciación hacia músculo estriado. Se origina a partir de células mesenquimales indiferenciadas dentro de la órbita y no es una transformación maligna de tejido muscular diferenciado como los músculos extraoculares. Es el tumor maligno orbitario más frecuente en niños y debe ser la primera consideración cuando se observa un tumor orbitario de rápida expansión en un niño.
En Estados Unidos, representa aproximadamente el 5% de todos los cánceres infantiles, con unos 250 casos nuevos diagnosticados anualmente. De estos, alrededor del 10% (aproximadamente 35 casos por año) son primarios orbitarios. La incidencia anual se estima en 4 casos por millón de habitantes en la población general y 4.5 casos por millón en niños 1).
La edad promedio de inicio es de 7 a 8 años, con dos tercios ocurriendo en niños menores de 10 años. El 90% ocurre en menores de 16 años, con un ligero predominio masculino (proporción hombre:mujer 5:3). No hay diferencia en la incidencia por raza. El tumor está bien delimitado y a menudo se localiza en la parte superior de la órbita.
La distribución del rabdomiosarcoma orbitario por sitio es: órbita 76%, conjuntiva 12%, úvea 9% y párpado 3%. Puede originarse dentro de la órbita o invadir la órbita desde tejidos circundantes como los senos paranasales. La órbita es el sitio primario en el 10% de todos los rabdomiosarcomas; otros sitios incluyen el tracto genitourinario (22%), extremidades (18%), región parameníngea (16%) y otras áreas de cabeza y cuello (10%).
Q¿A qué edad ocurre con mayor frecuencia el rabdomiosarcoma?
A
Dos tercios ocurren en niños menores de 10 años, con una edad promedio de inicio de 7 a 8 años. El 90% ocurre en menores de 16 años. La aparición en adultos es rara pero se ha reportado.
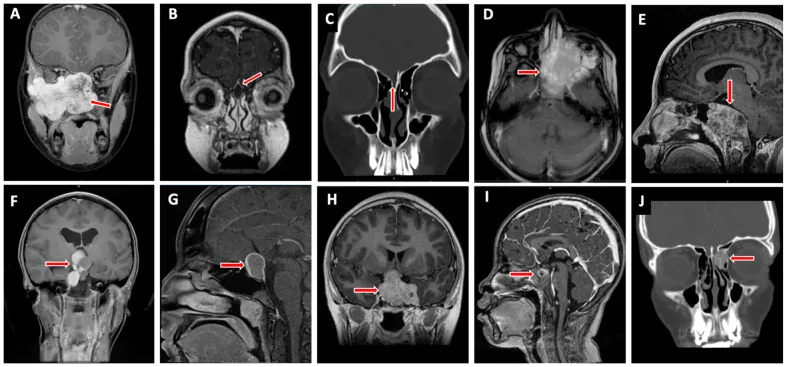
Valencia-Sanchez BA, et al. Special Considerations in Pediatric Endoscopic Skull Base Surgery. J Clin Med. 2024. Figure 2. PMCID: PMC11013018. License: CC BY.
La flecha roja indica rabdomiosarcoma (D), una de las lesiones representativas de la base del cráneo en niños. Esto corresponde a la infiltración tumoral tratada en la sección “2. Síntomas principales y hallazgos clínicos”.
Proptosis rápida: La proptosis unilateral rápidamente progresiva en cuestión de semanas es un síntoma típico.
Hinchazón palpebral: El párpado se hincha a medida que el tumor crece. Incluso con hinchazón severa, el enrojecimiento es leve, los hallazgos inflamatorios son escasos y el dolor no es intenso, lo cual es característico.
Se presentan diversas manifestaciones clínicas como limitación de la motilidad ocular y ptosis, según la ubicación del tumor.
Ocurre con frecuencia en niños de 0 a 9 años y se caracteriza por una progresión rápida.
Las lesiones se localizan con mayor frecuencia en la parte superior o superomedial, desplazando el ojo hacia abajo y hacia afuera. Se observa una masa redonda u ovalada, relativamente bien delimitada, fuera del cono muscular.
Aproximadamente el 10% de los casos se originan en los senos paranasales o la cavidad nasal e invaden secundariamente la órbita, pudiendo presentar sinusitis, congestión nasal o epistaxis. Los tumores posteriores pueden asociarse con pliegues coroideos, desprendimiento de retina o edema del disco óptico.
Las lesiones primarias conjuntivales aparecen como una masa granulomatosa rosada en forma de uva en el fondo de saco conjuntival. Las lesiones primarias uveales se presentan como masas del iris y pueden acompañarse de siembra en la cámara anterior o glaucoma secundario.
Las metástasis ocurren con mayor frecuencia en los pulmones, seguidas de la médula ósea, los huesos y los ganglios linfáticos. Aunque la órbita casi no tiene vasos linfáticos, los tumores anteriores de la conjuntiva y el párpado pueden metastatizar a los ganglios linfáticos regionales.
Q¿Qué enfermedades se confunden fácilmente con el rabdomiosarcoma?
A
Los diagnósticos diferenciales clínicos incluyen neuroblastoma, cloroma, linfangioma, hemangioma infantil, celulitis orbitaria e inflamación inespecífica. Además, la extensión orbitaria de sinusitis, rotura de quiste dermoide, sarcoma de Ewing e infiltración orbitaria de leucemia también se incluyen en el diagnóstico diferencial. La diferenciación se realiza mediante la progresión rápida, los hallazgos de imagen y los resultados de la biopsia.
El rabdomiosarcoma no se origina en los músculos extraoculares, sino en células mesenquimales indiferenciadas con potencial multilineal en los tejidos blandos orbitarios. La mayoría son esporádicos y se desconoce la causa exacta.
Los factores de riesgo genéticos asociados son los siguientes:
Neurofibromatosis tipo 1: Se sabe que está asociada con un mayor riesgo de desarrollar rabdomiosarcoma.
Síndrome de Li-Fraumeni: Implica anomalías en el gen supresor de tumores p53.
Síndrome de Beckwith-Wiedemann: Un trastorno congénito caracterizado por crecimiento excesivo, hernia umbilical e hipoglucemia.
Retinoblastoma hereditario: Puede ocurrir como un cáncer secundario después de la radioterapia.
Se realiza una TC de cabeza y órbita de urgencia para comprobar la presencia de un tumor intraorbitario, la homogeneidad del tumor, la destrucción de la pared ósea orbitaria y la extensión intracraneal o a los senos paranasales. A continuación, se realiza una RM para evaluar las características internas del tumor y su relación posicional con el globo ocular y los músculos extraoculares.
Los hallazgos característicos de cada modalidad son los siguientes:
Exploración
Hallazgos principales
TC
Masa homogénea, bien definida, redonda a ovalada. Realce moderado a intenso.
RM T1
Hipointensa respecto a la grasa orbitaria, isointensa respecto a los músculos extraoculares.
En la RM, la diferenciación del hemangioma capilar puede ser difícil, pero el hemangioma capilar se distingue por su rica vascularización (flujo sanguíneo). Cuando hay abundante componente estromal, la intensidad de señal en RM es diferente.
Para la búsqueda de metástasis se realizan radiografía de tórax, gammagrafía ósea y citología de aspiración de médula ósea. También se utilizan PET/TC, TC corporal total y gammagrafía para la evaluación sistémica.
Se necesita una biopsia para el diagnóstico definitivo. La biopsia por aspiración con aguja fina es insuficiente; se debe realizar una biopsia excisional o incisional.
Biopsia excisional: Se selecciona cuando es posible la extirpación quirúrgica sin dañar estructuras importantes.
Biopsia incisional: Se selecciona cuando el tumor es grande o está ubicado en la órbita posterior.
El diagnóstico intraoperatorio por congelación puede ser difícil de distinguir de otros tumores. El principio es realizar una biopsia mediante cirugía de urgencia e iniciar quimioterapia/radioterapia de forma temprana.
La demostración de rabdomioblastos mediante microscopía óptica, inmunohistoquímica y microscopía electrónica es fundamental para el diagnóstico.
Las estrías se detectan mediante tinción HE y tricrómica de Masson. La tinción inmunohistoquímica se utiliza para diferenciar células indiferenciadas, y un patrón de desmina positivo, HHF-35 (actina) positivo y α-actina de músculo liso negativo es útil para el diagnóstico. La microscopía electrónica revela haces de actina-miosina y proteínas de las bandas A, I y Z.
Diagnósticos diferenciales clínicos: neuroblastoma, cloroma, linfangioma, hemangioma infantil, celulitis orbitaria, inflamación inespecífica. Además, también se consideran en el diagnóstico diferencial la extensión orbitaria de sinusitis, celulitis orbitaria, rotura de quiste dermoide, hemorragia intratumoral de hemangioma, hemorragia de linfangioma, sarcoma de Ewing, infiltración orbitaria de leucemia (cloroma) y hemorragia orbitaria.
La resección quirúrgica completa no es posible y no debe ser el objetivo. Actualmente, el propósito principal de la cirugía es obtener un diagnóstico tisular, y la era de la exenteración orbitaria como primera opción ha terminado. Después de confirmar el diagnóstico patológico mediante biopsia por escisión, se realizan TC toracoabdominal y biopsia de médula ósea en el departamento de pediatría para verificar la presencia de metástasis sistémicas como en los pulmones. La quimioterapia sistémica es la base del tratamiento, y la combinación de cirugía, radioterapia y quimioterapia es el tratamiento estándar.
Para la quimioterapia, se utiliza principalmente la terapia VAC (vincristina + actinomicina D + ciclofosfamida). También se ha informado de los beneficios de ifosfamida y etopósido.
La radioterapia se administra a aproximadamente 40–60 Gy. Desde abril de 2016, la terapia con protones está cubierta por el seguro y se ha convertido en una opción de tratamiento estándar. La terapia con protones tiene la ventaja de reducir la dosis a los tejidos normales. El trasplante de células madre hematopoyéticas también se realiza en algunos casos.
Estadificación del IRS y estrategia de tratamiento
La estrategia de tratamiento para el rabdomiosarcoma orbitario se determina según el sistema de estadificación del Intergroup Rhabdomyosarcoma Study (IRS). En una serie de 30 casos de Shields et al., la distribución fue Grupo I 7%, Grupo II 37%, Grupo III 53% y Grupo IV 3%.
Grupo
Definición
Estrategia de tratamiento
I
Resección completa de la enfermedad localizada
Solo quimioterapia
II
Enfermedad residual o metástasis en ganglios linfáticos regionales
Quimioterapia + radioterapia
III
Resección incompleta o enfermedad residual macroscópica
Quimioterapia + radioterapia
IV
Metástasis a distancia
Quimioterapia + radioterapia
Para los grupos II a IV, la radioterapia se administra a 4000–5000 cGy (40–50 Gy) durante 4–5 semanas.
En casos que no pueden resecarse completamente o en recidivas, se considera tratamiento paliativo como radioterapia o quimioterapia. Los tratamientos quirúrgicos incluyen la extirpación del tumor y la exenteración orbitaria. Incluso si se logra una resección completa mediante cirugía, la quimioterapia es obligatoria. La órbita es un sitio de pronóstico favorable, y con el tratamiento adecuado se puede esperar una supervivencia superior al 90%.
Si hay invasión intracraneal o de los senos paranasales, o metástasis en pulmones, otras partes del cuerpo o ganglios linfáticos cervicales, el pronóstico es malo. La detección temprana y el inicio temprano de la quimioterapia conducen a una buena respuesta y mejor pronóstico.
Las complicaciones oftálmicas de la quimioterapia incluyen queratoconjuntivitis seca y blefaroconjuntivitis por ciclofosfamida, conjuntivitis y visión borrosa por ifosfamida, y oclusión de la arteria central de la retina por etopósido.
La recurrencia local se evalúa mediante resonancias magnéticas periódicas. La metástasis hematógena es frecuente, por lo que se realizan exámenes sistémicos regulares.
Los resultados visuales a largo plazo después del tratamiento (Shields) son: 20/20 a 20/40 en el 39%, 20/50 a 20/100 en el 18%, y 20/200 a sin percepción de luz en el 43%.
Q¿Qué complicaciones oculares pueden ocurrir después del tratamiento del rabdomiosarcoma?
El rabdomiosarcoma se clasifica según la OMS en cuatro subtipos: embrionario, alveolar, pleomórfico y de células fusiformes/esclerosante 2).
Tipo embrionario
Frecuencia: Representa el 50–60% de todos los rabdomiosarcomas y el 80–84% de los rabdomiosarcomas orbitarios.
Histología: Haces de células fusiformes (células vellosas) que discurren en varias direcciones. Las células son pequeñas redondas o fusiformes con escaso pleomorfismo nuclear. El citoplasma es escaso con gránulos eosinófilos, y se observan rabdomioblastos con estrías transversales. Más frecuente en la órbita inferior.
Pronóstico: La tasa de supervivencia a 5 años para los casos orbitarios es del 95%, lo cual es favorable. La edad media de aparición es de 7 a 8 años.
Tipo Alveolar
Frecuencia: Representa aproximadamente el 20% de todos los rabdomiosarcomas y alrededor del 10% de los rabdomiosarcomas orbitarios.
Histología: El tumor crece en un patrón alveolar. Células grandes e irregulares con núcleos grandes y citoplasma eosinófilo abundante se disponen alrededor de tabiques fibrosos. Propenso a metástasis extensas.
Pronóstico: La tasa de supervivencia a 5 años para los casos orbitarios es del 74%. Es el tipo histológico más maligno y de peor pronóstico.
Tipo Pleomórfico (Tipo Diferenciado)
Frecuencia: Raro en niños. Ocurre principalmente en las extremidades (especialmente el muslo) de adultos.
Histología: Células redondas a alargadas con multinucleación y citoplasma abundante, con estrías transversales identificables. Compuesto por células tumorales altamente pleomórficas. Baja frecuencia pero alta diferenciación.
Pronóstico: Relativamente favorable en casos orbitarios.
El tipo botrioide es una variante del tipo embrionario común en lactantes, caracterizado por agregados de células tumorales subepiteliales que dan una apariencia “similar a un racimo de uvas”.
Histológicamente, los tipos alveolar y pleomórfico tienen peor pronóstico que el tipo embrionario. La tasa de supervivencia global a 5 años para el rabdomiosarcoma es del 66%, pero el rabdomiosarcoma orbitario se clasifica como un grupo de pronóstico favorable según su sitio de origen.
El rabdomiosarcoma alveolar se caracteriza por translocaciones cromosómicas recurrentes t(2;13)(q35;q14) y t(1;13)(p36;q14). Aproximadamente el 80% de los rabdomiosarcomas alveolares involucran translocaciones PAX3-FOXO1 o PAX7-FOXO1. La fusión PAX3-FOXO1 se asocia con alta expresión de OLIG2 y mal pronóstico2).
El rabdomiosarcoma embrionario no presenta reordenamientos cromosómicos estructurales recurrentes, pero se observa con frecuencia pérdida alélica en el cromosoma 11 (especialmente la región 11p15.5).
Q¿Cuál es la diferencia en el pronóstico entre los tipos embrionario y alveolar?
A
La tasa de supervivencia a 5 años para el tipo embrionario en el rabdomiosarcoma orbitario es del 95%, mientras que para el tipo alveolar es más baja, del 74%. El tipo alveolar se asocia con translocaciones cromosómicas como PAX3-FOXO1 y es un tipo histológico propenso a metástasis extensas.
7. Investigación más reciente y perspectivas futuras (informes en fase de investigación)
En los últimos años, se ha identificado un nuevo subtipo llamado rabdomiosarcoma de células fusiformes epitelioides con reordenamiento de TFCP2.
Li et al. (2023) informaron que el rabdomiosarcoma de células fusiformes epitelioides ocurre preferentemente en huesos (cabeza y cuello, pelvis) y se caracteriza por fusiones EWSR1-TFCP2 o FUS-TFCP2, representando un subtipo de pronóstico extremadamente malo3). La supervivencia media de los casos reportados es de solo 17 meses.
Se están llevando a cabo investigaciones terapéuticas dirigidas a la proteína de fusión PAX3-FOXO1, que está implicada en aproximadamente el 80% de los rabdomiosarcomas alveolares.
Se ha informado que una formulación que encapsula ARN pequeño de interferencia (siRNA) en partículas de liposoma-protamina regula a la baja eficientemente la expresión de PAX3-FOXO1 en líneas celulares de rabdomiosarcoma alveolar in vitro y retrasa o suprime el crecimiento de tumores de rabdomiosarcoma alveolar en xenoinjertos1).
Se está intentando la aplicación de inhibidores de puntos de control inmunitario (como nivolumab) en sarcomas metastásicos1). Sin embargo, en la actualidad no se ha establecido su eficacia en el rabdomiosarcoma.
Tratamiento del rabdomiosarcoma primario en adultos
Una revisión bibliográfica sobre el rabdomiosarcoma alveolar primario de la región pineal en adultos sugiere una asociación entre la intensidad del tratamiento y la supervivencia.
En una revisión de 13 casos de rabdomiosarcoma alveolar primario de la región pineal en adultos realizada por Chang et al. (2025), la supervivencia media fue de aproximadamente 5 meses solo con cirugía, alrededor de 10.28 meses con cirugía más radioterapia, y aproximadamente 11.33 meses con cirugía más radioterapia más quimioterapia, lo que indica una tendencia a una supervivencia prolongada con el tratamiento multimodal2).
Yang N, Kong D, Wang X, Liu Y. Perianal rhabdomyosarcoma in an adult: A case report and review of the literature. Medicine. 2023;102(48):e36199.
Chang T, Ding C, Liu Y, Yang Y, Mao Q.. Primary pineal alveolar rhabdomyosarcoma in an adult patient: a case report and literature review. BMC Neurol. 2025;25(1):106. doi:10.1186/s12883-025-04113-8. PMID:40082819; PMCID:PMC11905731.
Li Y, Li D, Wang J, Tang J.. Epithelioid and spindle rhabdomyosarcoma with TFCP2 rearrangement in abdominal wall: a distinctive entity with poor prognosis. Diagn Pathol. 2023;18(1):41. doi:10.1186/s13000-023-01330-y. PMID:36998041; PMCID:PMC10061849.
Copia el texto del artículo y pégalo en el asistente de IA que prefieras.
Artículo copiado al portapapeles
Abre un asistente de IA abajo y pega el texto copiado en el chat.