El glioma del nervio óptico (glioma del nervio óptico / glioma de la vía óptica) es un tipo de glioma que se origina en el nervio óptico. En sentido estricto, se refiere a un glioma que surge en el nervio óptico antes del quiasma óptico. En sentido amplio, significa un glioma que surge en toda la vía óptica, incluyendo la región posterior al quiasma óptico (glioma de la vía óptica).
Histológicamente, la mayoría son astrocitomas pilocíticos benignos (astrocitoma pilocítico, Grado I de la OMS). Sin embargo, también se han reportado algunos casos malignos. Aproximadamente el 70% se presenta en la infancia, y es una enfermedad rara que representa alrededor del 0.5 al 5% de los tumores cerebrales pediátricos.
Existe una fuerte asociación con la neurofibromatosis tipo 1 (NF1, enfermedad de von Recklinghausen), y se observa NF1 concomitante en aproximadamente el 20-30% de los casos de glioma del nervio óptico. Por el contrario, el glioma del nervio óptico es la lesión orbitaria más común en pacientes con NF1.
Clasificación según la localización y el antecedente genético
Forma de presentación más frecuente. Los síntomas principales son pérdida visual unilateral y proptosis.
Se limita al nervio óptico dentro de la órbita, y la observación es la base del manejo.
En casos asociados con NF1, se ha reportado regresión espontánea.
Tipo infiltrante del quiasma óptico
Tipo que infiltra hasta el quiasma óptico.
Causa discapacidad visual binocular y su manejo es complejo.
Ocurre con frecuencia en niños pequeños; se debe evaluar la extensión al hipotálamo.
Tipo quiasmático-hipotalámico
Tipo que se extiende desde la parte posterior del quiasma hasta el hipotálamo.
Puede asociarse con anomalías endocrinas (trastornos del crecimiento, pubertad precoz, etc.).
El tratamiento requiere colaboración entre neurocirugía y endocrinología.
En cuanto a la clasificación según antecedentes genéticos, existen el tipo asociado a NF1 (aproximadamente el 30% del total) y el tipo esporádico (aproximadamente el 70%).
En el tipo asociado a NF1 también se observa afectación bilateral.
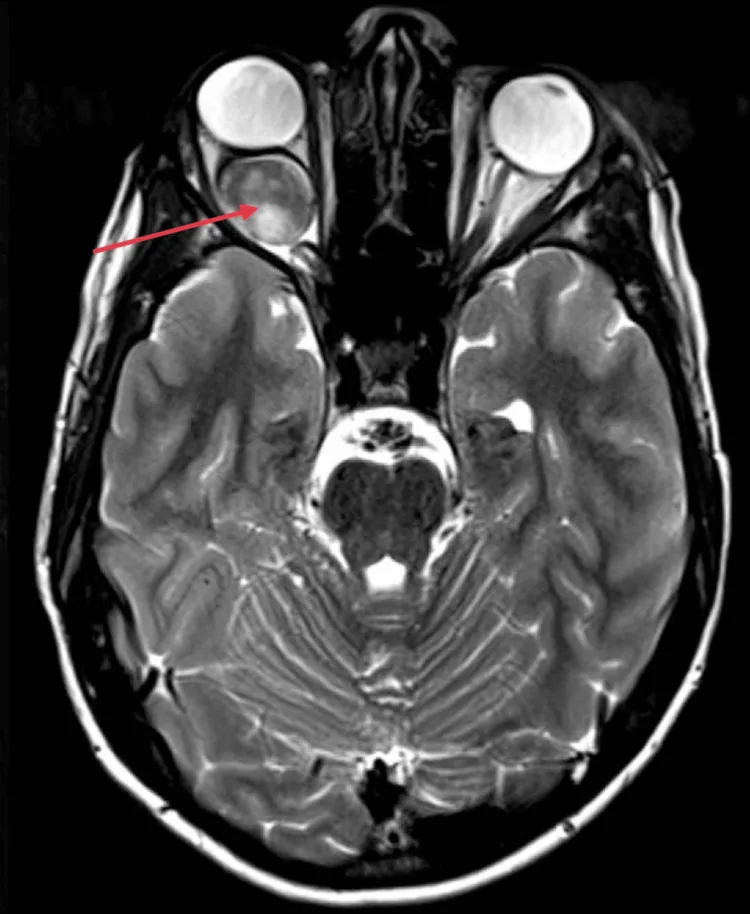
Kotecha MR, et al. Idiopathic Optic Nerve Glioma: A Case Report. Cureus. 2024. Figure 2. PMCID: PMC11744336. License: CC BY.
En la RM axial, el nervio óptico intraorbitario derecho muestra un agrandamiento fusiforme con realce homogéneo e intenso (flecha roja), y se confirma proptosis debido al efecto de masa hacia la parte posterior del globo ocular. Corresponde a los hallazgos de imagen por RM (agrandamiento uniforme del nervio óptico y curvatura descendente) tratados en la sección «2. Principales síntomas y hallazgos clínicos».
Los niños pequeños no se quejan de la disminución de la visión. Por lo tanto, a menudo son los padres o las personas cercanas quienes notan el estrabismo (especialmente la endotropía) y acuden por primera vez al oftalmólogo.
La disminución de la visión monocular sin estrabismo es aún más difícil de detectar. En la primera consulta, ya puede haber atrofia del nervio óptico.
El glioma del nervio óptico bilateral es más común en casos de inicio temprano. Se nota por primera vez por anomalías en la mirada o comportamientos de “no ver”, y la discapacidad visual puede ser grave.
La proptosis no es evidente y no hay dolor.
QSi a un niño le diagnostican estrabismo, ¿es posible que tenga un glioma del nervio óptico?
A
En el glioma del nervio óptico, los niños pequeños a menudo no pueden notar ni quejarse de la disminución de la visión, por lo que el estrabismo (especialmente la endotropía) suele ser el síntoma inicial que lleva a la consulta oftalmológica. En niños diagnosticados con estrabismo, especialmente si es unilateral, es importante considerar la posibilidad de un glioma del nervio óptico y realizar una evaluación completa que incluya agudeza visual, fondo de ojo y pruebas de imagen.
En las lesiones orbitarias de NF1, el glioma óptico es el más común
Q¿Es más probable desarrollar un glioma del nervio óptico si se tiene NF1 (neurofibromatosis tipo 1)?
A
Los pacientes con NF1 tienen un riesgo significativamente mayor de glioma del nervio óptico, y entre el 20 y el 30% de todos los casos de glioma del nervio óptico están asociados con NF11). Tras el diagnóstico de NF1, se recomienda un seguimiento oftalmológico periódico como cribado del glioma del nervio óptico. Por el contrario, si se descubre un glioma del nervio óptico en un niño, es importante verificar en todos los casos si se cumplen los criterios diagnósticos de NF1.
Enfermedad más importante en el diagnóstico diferencial.
Es más frecuente en mujeres adultas y puede asociarse con NF2.
En CT/RM, el signo del tranvía (tram-track sign) es característico y útil para diferenciarlo del glioma del nervio óptico.
Neuritis óptica
Suele presentarse con inicio agudo y dolor con los movimientos oculares.
La RM muestra realce del nervio óptico con contraste, pero el aumento de tamaño es leve.
Suele mejorar con el tratamiento con esteroides.
El glioma del nervio óptico muestra un agrandamiento uniforme y una incurvación hacia abajo (downward kinking), que se distingue claramente del signo de la vía del tranvía (tram-track sign) del meningioma de la vaina del nervio óptico.
Q¿Cuál es la diferencia entre el glioma del nervio óptico y el meningioma de la vaina del nervio óptico?
A
El glioma del nervio óptico es un tumor benigno (astrocitoma pilocítico) frecuente en niños, a menudo asociado con NF1. En la TC/RM se caracteriza por un agrandamiento uniforme del nervio óptico y una incurvación hacia abajo (downward kinking). El meningioma de la vaina del nervio óptico es más común en mujeres adultas, puede asociarse con NF2, y en la TC/RM presenta el signo de la vía del tranvía (tram-track sign: calcificación o realce de contraste a lo largo de la vaina del nervio óptico), lo que permite diferenciarlos.
Dado que el tumor es benigno y frecuente en niños, si está limitado al nervio óptico intraorbitario, en principio no se realiza resección quirúrgica ni radioterapia. La estrategia básica es una observación cuidadosa con imágenes periódicas (RM cada 3 a 6 meses).
Antiguamente se realizaba cirugía, pero debido al alto riesgo de ceguera irreversible, actualmente se tiende a evitar la resección quirúrgica. En casos asociados a NF1, se ha reportado regresión espontánea, por lo que se realiza una observación especialmente cuidadosa.
Quimioterapia (casos progresivos o con pérdida visual)
En casos de pérdida visual progresiva o crecimiento tumoral, se utiliza quimioterapia combinada con carboplatino y vincristina (régimen CV) como tratamiento de primera línea estándar3)4).
Régimen estándar de CV (COG A9952, etc.):
Vincristina: 1.5 mg/m² IV, una vez por semana × 10 semanas
Carboplatino: 550 mg/m² IV, cada 3 semanas
La tasa de respuesta objetiva (remisión parcial + estabilización) con CV se reporta en 60-80%4).
Opciones de segunda línea:
Cisplatino + etopósido
Temozolomida (agente alquilante)
Vinblastina en monoterapia (tras recurrencia a terapia con CV5))
Se considera en casos avanzados resistentes a la quimioterapia. Sin embargo, en niños se tiende a evitar en lo posible debido al riesgo de cáncer secundario, disfunción endocrina (por irradiación cercana al hipotálamo) y efectos sobre la función cognitiva.
El hallazgo histopatológico del glioma del nervio óptico es un astrocitoma pilocítico benigno (astrocitoma pilocítico, grado I de la OMS). Es esencialmente diferente de los gliomas de alto grado, como el glioblastoma multiforme (grado IV de la OMS).
Las células tumorales muestran una morfología característica con prolongaciones celulares bipolares y contienen fibras de Rosenthal. Se originan de las células gliales (astrocitos) del nervio óptico y comprimen y reemplazan el nervio óptico desde el interior.
El glioma del nervio óptico asociado a NF1 (neurofibromatosis tipo 1) es causado por mutaciones en el gen NF1 (cromosoma 17q11.2).
El gen NF1 es un gen supresor de tumores que codifica la proteína neurofibromina.
neurofibromina funciona como proteína activadora de Ras-GTPasa (GAP) y suprime la señalización de Ras
Mutación de NF1 → pérdida de función de neurofibromina → activación constitutiva de la vía de señalización de Ras → aumento de la señalización MAPK → proliferación descontrolada de células gliales
En el astrocitoma pilocítico esporádico (no asociado a NF1), se observa con frecuencia el gen de fusión BRAF-KIAA1549. Este gen de fusión también activa la vía MAPK y promueve el crecimiento tumoral.
Algunos casos presentan la mutación BRAF V600E, y aquellos que la tienen tienden a ser de mayor grado de malignidad6).
La mayoría de los gliomas del nervio óptico son de bajo grado (low-grade) y muestran un crecimiento lento.
El tumor agranda el nervio óptico desde el interior y provoca una flexión (kinking o downward kinking) del nervio dentro de la órbita.
El agrandamiento uniforme y la flexión hacia abajo en la RM son puntos clave para el diagnóstico por imagen.
El pronóstico visual no es uniforme; dado que coexisten casos progresivos y estables, se requiere una evaluación longitudinal de la función visual además del tamaño del tumor. Existen casos en los que la función visual empeora a pesar de hallazgos estables en la RM, y, por el contrario, en pacientes con NF1 puede observarse regresión espontánea. 1, 8, 9)
Evaluación de la función visual: medir repetidamente la agudeza visual, campo visual, visión cromática y RAPD según la edad
Seguimiento por imagen: mediante RMN para detectar recrecimiento, extensión quiasmática e intracraneal8)
Evaluación endocrina: en casos de extensión hipotalámica, confirmar trastornos del crecimiento, pubertad precoz y diabetes insípida
Manejo sistémico de NF1: realizar seguimiento integral que incluya lesiones cutáneas, otros tumores y aspectos del desarrollo2, 9)
Se ha informado la eficacia de los inhibidores de MEK en gliomas de bajo grado asociados a NF1.
En el ensayo SPRINT (Fase II), se reportó una tasa de respuesta objetiva del 66% con selumetinib en gliomas de bajo grado progresivos asociados a NF1 (neurofibromas plexiformes)7). Se está evaluando su aplicación en gliomas de bajo grado asociados a NF1, incluidos los gliomas del nervio óptico.
Para gliomas de bajo grado pediátricos con mutación BRAF V600E, la terapia combinada de dabrafenib más trametinib se está evaluando en ensayos clínicos6). En casos con fusión BRAF-KIAA1549 positiva, la eficacia de los inhibidores de BRAF es limitada.
Dirección de los fármacos de terapia dirigida molecular
Con la llegada de los inhibidores de MEK y BRAF, se está produciendo un cambio desde la quimioterapia convencional (régimen CV) hacia un tratamiento personalizado basado en el perfil molecular8). En el futuro, es posible que la selección del tratamiento se estandarice según el perfil de mutaciones genéticas (fusión BRAF, BRAF V600E, mutación NF1, etc.).
Listernick R, Charrow J, Greenwald M, Mets M. Natural history of optic pathway tumors in children with neurofibromatosis type 1: a longitudinal study. J Pediatr. 1994;125(1):63-66. doi:10.1016/s0022-3476(94)70122-9. PMID:8021787.
Ferner RE, Ferner RE, Huson SM, Thomas N, Moss C, Willshaw H, Evans DG, Upadhyaya M, Towers R, Gleeson M, Steiger C, Kirby A. Guidelines for the diagnosis and management of individuals with neurofibromatosis 1. J Med Genet. 2007;44(2):81-88. doi:10.1136/jmg.2006.045906. PMID:17105749; PMCID:PMC2598063.
Gnekow AK, Walker DA, Kandels D, Picton S, Giorgio Perilongo, Grill J, Stokland T, Sandstrom PE, Warmuth-Metz M, Pietsch T, Giangaspero F, Schmidt R, Faldum A, Kilmartin D, De Paoli A, De Salvo GL, of the Low Grade Glioma Consortium and the participating centers. A European randomised controlled trial of the addition of etoposide to standard vincristine and carboplatin induction as part of an 18-month treatment programme for childhood (≤16 years) low grade glioma - A final report. Eur J Cancer. 2017;81:206-225. doi:10.1016/j.ejca.2017.04.019. PMID:28649001; PMCID:PMC5517338.
Ater JL, Zhou T, Holmes E, Mazewski CM, Booth TN, Freyer DR, Lazarus KH, Packer RJ, Prados M, Sposto R, Vezina G, Wisoff JH, Pollack IF. Randomized study of two chemotherapy regimens for treatment of low-grade glioma in young children: a report from the Children’s Oncology Group. J Clin Oncol. 2012;30(21):2641-2647. doi:10.1200/jco.2011.36.6054. PMID:22665535; PMCID:PMC3413276.
Alvaro Lassaletta, Katrin Scheinemann, Shayna M. Zelcer, Juliette Hukin, Beverley A. Wilson, Nada Jabado, Anne Sophie Carret, Lucie Lafay-Cousin, et al. Phase II Weekly Vinblastine for Chemotherapy-Naïve Children With Progressive Low-Grade Glioma: A Canadian Pediatric Brain Tumor Consortium Study. JCO. 2016;34(29):3537-3543. doi:10.1200/jco.2016.68.1585.
Fangusaro J, Onar-Thomas A, Young Poussaint T, Wu S, Ligon AH, Lindeman N, Banerjee A, Packer RJ, Kilburn LB, Goldman S, Pollack IF, Qaddoumi I, Jakacki RI, Fisher PG, Dhall G, Baxter P, Kreissman SG, Stewart CF, Jones DTW, Pfister SM, Vezina G, Stern JS, Panigrahy A, Patay Z, Tamrazi B, Jones JY, Haque SS, Enterline DS, Cha S, Fisher MJ, Doyle LA, Smith M, Dunkel IJ, Fouladi M. Selumetinib in paediatric patients with BRAF-aberrant or neurofibromatosis type 1-associated recurrent, refractory, or progressive low-grade glioma: a multicentre, phase 2 trial. Lancet Oncol. 2019;20(7):1011-1022. doi:10.1016/s1470-2045(19)30277-3. PMID:31151904; PMCID:PMC6628202.
Anuradha Banerjee, Regina I. Jakacki, Arzu Onar-Thomas, Shengjie Wu, Theodore Nicolaides, Tina Young Poussaint, Jason Fangusaro, Joanna Phillips, et al. A phase I trial of the MEK inhibitor selumetinib (AZD6244) in pediatric patients with recurrent or refractory low-grade glioma: a Pediatric Brain Tumor Consortium (PBTC) study. Neuro-Oncology. 2017;19(8):1135-1144. doi:10.1093/neuonc/now282.
de Blank PMK, Orne-Ibekwe E, Packer R. International consensus recommendations for visual surveillance in optic pathway gliomas associated with neurofibromatosis type 1. J Neurooncol. 2020;148(3):571-578.
Listernick R, Louis DN, Packer RJ, Gutmann DH. Optic pathway gliomas in children with neurofibromatosis 1: consensus statement from the NF1 Optic Pathway Glioma Task Force. Ann Neurol. 1997;41(2):143-149. doi:10.1002/ana.410410204. PMID:9029062.
Copia el texto del artículo y pégalo en el asistente de IA que prefieras.
Artículo copiado al portapapeles
Abre un asistente de IA abajo y pega el texto copiado en el chat.