Le gliome du nerf optique (optic nerve glioma / optic pathway glioma) est un type de gliome qui se développe sur le nerf optique. Au sens strict, il désigne un gliome situé sur le nerf optique avant le chiasma optique. Au sens large, il désigne un gliome situé sur l’ensemble de la voie optique, y compris en arrière du chiasma (optic pathway glioma).
Histologiquement, il s’agit le plus souvent d’un astrocytome pilocytique bénin (pilocytic astrocytoma, WHO Grade I). Cependant, des cas malins ont également été rapportés. Environ 70 % des cas surviennent dans l’enfance, et il s’agit d’une maladie rare représentant environ 0,5 à 5 % des tumeurs cérébrales pédiatriques.
Il existe une forte association avec la neurofibromatose de type 1 (NF1, maladie de von Recklinghausen), et environ 20 à 30 % des cas de gliome du nerf optique présentent une NF1 associée. Inversement, le gliome du nerf optique est la lésion orbitaire la plus fréquente chez les patients atteints de NF1.
Classification selon la localisation et le contexte génétique
Forme la plus fréquente. Les principaux symptômes sont une baisse de vision unilatérale et une exophtalmie.
Limitée au nerf optique intra-orbitaire, la surveillance est la règle. Une régression spontanée a été rapportée dans les cas associés à la NF1.
Forme avec infiltration chiasmatique
Forme qui infiltre le chiasma optique.
Il entraîne une perte de vision bilatérale et une prise en charge complexe.
Il survient souvent à un âge précoce ; il faut évaluer l’extension vers l’hypothalamus.
Type chiasmatique-hypothalamique
Type s’étendant de l’arrière du chiasma à l’hypothalamus.
Des anomalies endocriniennes (troubles de la croissance, puberté précoce, etc.) peuvent être associées.
Sur le plan thérapeutique, une collaboration avec la neurochirurgie et l’endocrinologie est nécessaire.
La classification selon le contexte génétique distingue le type associé à la NF1 (environ 30 % des cas) et le type sporadique (environ 70 %).
Dans le type associé à la NF1, une atteinte bilatérale peut également survenir.
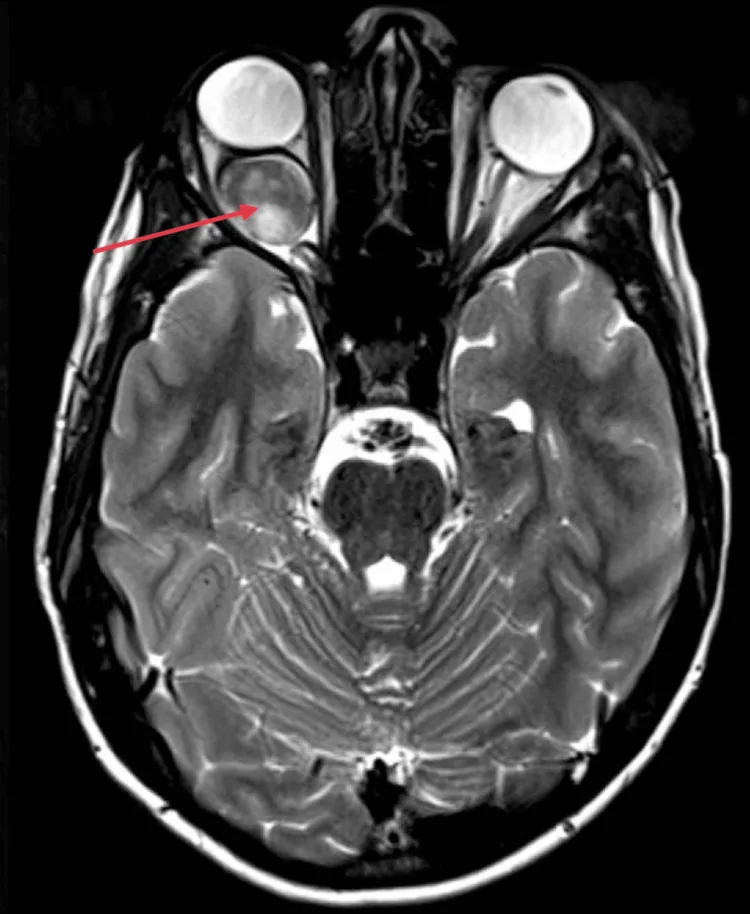
Kotecha MR, et al. Idiopathic Optic Nerve Glioma: A Case Report. Cureus. 2024. Figure 2. PMCID: PMC11744336. License: CC BY.
L’IRM axiale montre un élargissement fusiforme du nerf optique dans l’orbite droite, avec un rehaussement intense et homogène (flèche rouge), et une exophtalmie due à l’effet de masse postérieur au globe oculaire. Cette image correspond aux signes IRM (élargissement homogène du nerf optique, coudure vers le bas) traités dans la section « 2. Principaux symptômes et signes cliniques ».
Les jeunes enfants ne se plaignent pas spontanément d’une baisse de vision. Par conséquent, ce sont souvent les parents ou les personnes de l’entourage qui remarquent un strabisme (en particulier une ésotropie) et consultent un ophtalmologiste pour la première fois.
Une baisse de vision monoculaire sans strabisme est encore plus susceptible d’être détectée tardivement. Une atrophie optique peut déjà être présente lors de la première consultation.
Les gliomes optiques bilatéraux sont fréquents chez les enfants d’âge précoce. Ils sont souvent découverts pour la première fois en raison d’anomalies du regard ou de « comportements de non-voyance », et la déficience visuelle peut être grave.
L’exophtalmie n’est pas évidente et il n’y a pas de douleur.
QSi un enfant est diagnostiqué avec un strabisme, est-ce possible qu'il s'agisse d'un gliome du nerf optique ?
A
Dans le gliome du nerf optique, les jeunes enfants ne peuvent pas ressentir ou signaler une baisse de vision, donc le strabisme (en particulier l’ésotropie) est souvent le premier symptôme qui les amène à consulter un ophtalmologiste. Chez les enfants diagnostiqués avec un strabisme, en particulier un strabisme unilatéral, il est important d’envisager un gliome du nerf optique et de réaliser des examens approfondis incluant l’acuité visuelle, le fond d’œil et l’imagerie.
Le gliome du nerf optique est la lésion orbitaire la plus fréquente dans la NF1
QLes personnes atteintes de NF1 (neurofibromatose de type 1) sont-elles plus susceptibles de développer un gliome du nerf optique ?
A
Les patients atteints de NF1 présentent un risque considérablement accru de gliome du nerf optique, et 20 à 30 % de tous les cas de gliome du nerf optique sont associés à la NF11). Après le diagnostic de NF1, un suivi ophtalmologique régulier est recommandé pour le dépistage du gliome du nerf optique. Inversement, lorsqu’un gliome du nerf optique est découvert chez un enfant, il est important de vérifier systématiquement s’il répond aux critères diagnostiques de la NF1.
Baisse de vision unilatérale chez l’enfant, exophtalmie (même non évidente)
TDM/IRM : élargissement homogène du nerf optique et coudure vers le bas (downward kinking)
Vérifier systématiquement la présence d’une NF1 associée
La biopsie n’est généralement pas nécessaire (le diagnostic peut être posé par imagerie). La confirmation tissulaire est effectuée lors de l’exérèse chirurgicale.
Plus fréquent chez les femmes adultes, avec des cas associés à la NF2.
Le tram-track sign (signe de la voie ferrée) est caractéristique au CT/IRM, utile pour différencier du gliome du nerf optique.
Névrite optique
Début aigu, souvent accompagné de douleur lors des mouvements oculaires.
L’IRM montre un rehaussement du nerf optique après injection de contraste, mais l’élargissement est léger. L’amélioration est fréquente avec un traitement par corticoïdes.
Le gliome du nerf optique présente un élargissement homogène et un downward kinking, ce qui le distingue clairement du tram-track sign du méningiome de la gaine du nerf optique.
QQuelle est la différence entre le gliome du nerf optique et le méningiome de la gaine du nerf optique ?
A
Le gliome du nerf optique est une tumeur bénigne (astrocytome pilocytique) fréquente chez l’enfant, souvent associée à la NF1. Au scanner/IRM, il se caractérise par un élargissement homogène du nerf optique et un downward kinking. Le méningiome de la gaine du nerf optique touche davantage les femmes adultes, parfois associé à la NF2, et se distingue par un tram-track sign (calcification ou rehaussement linéaire le long du nerf optique) au scanner/IRM.
La tumeur étant bénigne et fréquente chez l’enfant, l’exérèse chirurgicale et la radiothérapie ne sont généralement pas pratiquées lorsqu’elle est limitée au nerf optique intra-orbitaire. La stratégie de base consiste en une surveillance attentive, principalement par imagerie par résonance magnétique (IRM) tous les 3 à 6 mois.
Autrefois, la chirurgie était pratiquée, mais en raison du risque élevé de cécité irréversible, on tend désormais à éviter l’exérèse chirurgicale. Des cas de régression spontanée ont été rapportés chez les patients atteints de NF1, justifiant une surveillance particulièrement prudente.
Chimiothérapie (cas évolutifs ou avec baisse de l’acuité visuelle)
En cas de baisse de l’acuité visuelle ou de progression tumorale, une chimiothérapie combinée par carboplatine et vincristine (protocole CV) est utilisée comme traitement de première ligne standard3)4).
Schéma standard de la thérapie CV (COG A9952, etc.) :
Vincristine : 1,5 mg/m² IV, une fois par semaine × 10 semaines
Carboplatine : 550 mg/m² IV, toutes les 3 semaines
Le taux de réponse objective (réponse partielle + stabilisation) de la thérapie CV est rapporté à 60–80 % 4).
Options de traitement de deuxième ligne :
Cisplatine + étoposide
Témozolomide (agent alkylant)
Vinblastine en monothérapie (récidive après traitement par CV5))
Envisagée en cas de progression malgré la chimiothérapie. Cependant, chez l’enfant, en raison des risques de second cancer, de dysfonction endocrinienne (irradiation proche de l’hypothalamus) et d’impact sur les fonctions cognitives, on tend à l’éviter autant que possible.
Les résultats histopathologiques du gliome du nerf optique sont ceux d’un astrocytome pilocytique bénin (astrocytome pilocytique, grade I de l’OMS). Il diffère fondamentalement des gliomes de haut grade comme le glioblastome multiforme (grade IV de l’OMS).
Les cellules tumorales présentent une morphologie caractéristique avec des prolongements cellulaires bipolaires et contiennent des fibres de Rosenthal. Elles proviennent des cellules gliales (astrocytes) du nerf optique, qu’elles compriment et remplacent de l’intérieur.
Le gène NF1 est un gène suppresseur de tumeur codant pour la protéine neurofibromine.
La neurofibromine fonctionne comme une protéine activatrice de la GTPase Ras (GAP) et supprime la signalisation Ras.
Mutation NF1 → perte de fonction de la neurofibromine → activation constitutive de la voie de signalisation Ras → augmentation de la signalisation MAPK → prolifération incontrôlée des cellules gliales.
Dans les astrocytomes pilocytiques sporadiques (non associés à NF1), le gène de fusion BRAF-KIAA1549 est fréquemment observé. Ce gène de fusion active également la voie MAPK et favorise la croissance tumorale.
Certains cas présentent une mutation BRAF V600E, et ceux-ci ont tendance à être de plus haut grade de malignité6).
La grande majorité des gliomes du nerf optique sont de bas grade et présentent une croissance lente.
La tumeur élargit le nerf optique de l’intérieur et provoque une coudure (kinking, downward kinking) du nerf dans l’orbite.
L’élargissement homogène et la coudure vers le bas à l’IRM sont des points clés du diagnostic radiologique.
Le pronostic visuel n’est pas uniforme, avec des cas d’aggravation et de stabilisation coexistants. Par conséquent, une évaluation longitudinale de la fonction visuelle est nécessaire, au-delà de la seule taille de la tumeur. Certains cas présentent une détérioration visuelle malgré des résultats IRM stables, tandis que d’autres, notamment dans le cadre de la NF1, peuvent montrer une régression spontanée. 1, 8, 9)
Évaluation visuelle : mesurer de manière répétée l’acuité visuelle, le champ visuel, la vision des couleurs et le RAPD en fonction de l’âge
Suivi d’imagerie : surveiller la récidive, l’extension chiasmatique et intracrânienne par IRM8)
Évaluation endocrinienne : en cas d’extension hypothalamique, vérifier les troubles de croissance, la puberté précoce et le diabète insipide
Prise en charge globale de la NF1 : assurer un suivi systémique incluant les lésions cutanées, les autres tumeurs et le développement2, 9)
L’efficacité des inhibiteurs de MEK pour les gliomes de bas grade associés à la NF1 a été rapportée.
Dans l’essai SPRINT (Phase II), un taux de réponse objective de 66 % a été rapporté pour le sélumétinib dans les gliomes de bas grade progressifs associés à la NF1 (neurofibromes plexiformes) 7). L’application aux gliomes de bas grade associés à la NF1, y compris les gliomes des voies optiques, est à l’étude.
Pour les gliomes de bas grade pédiatriques porteurs de la mutation BRAF V600E, la thérapie combinée dabrafénib + tramétinib est évaluée dans des essais cliniques6). Pour les cas positifs pour la fusion BRAF-KIAA1549, l’efficacité des inhibiteurs de BRAF est limitée.
Avec l’avènement des inhibiteurs de MEK et de BRAF, on observe un passage de la chimiothérapie conventionnelle (CV) vers un traitement personnalisé basé sur le profil moléculaire8). À l’avenir, la sélection thérapeutique basée sur le profil de mutation génétique (fusion BRAF, BRAF V600E, mutation NF1, etc.) pourrait devenir standardisée.
Listernick R, Charrow J, Greenwald M, Mets M. Natural history of optic pathway tumors in children with neurofibromatosis type 1: a longitudinal study. J Pediatr. 1994;125(1):63-66. doi:10.1016/s0022-3476(94)70122-9. PMID:8021787.
Ferner RE, Ferner RE, Huson SM, Thomas N, Moss C, Willshaw H, Evans DG, Upadhyaya M, Towers R, Gleeson M, Steiger C, Kirby A. Guidelines for the diagnosis and management of individuals with neurofibromatosis 1. J Med Genet. 2007;44(2):81-88. doi:10.1136/jmg.2006.045906. PMID:17105749; PMCID:PMC2598063.
Gnekow AK, Walker DA, Kandels D, Picton S, Giorgio Perilongo, Grill J, Stokland T, Sandstrom PE, Warmuth-Metz M, Pietsch T, Giangaspero F, Schmidt R, Faldum A, Kilmartin D, De Paoli A, De Salvo GL, of the Low Grade Glioma Consortium and the participating centers. A European randomised controlled trial of the addition of etoposide to standard vincristine and carboplatin induction as part of an 18-month treatment programme for childhood (≤16 years) low grade glioma - A final report. Eur J Cancer. 2017;81:206-225. doi:10.1016/j.ejca.2017.04.019. PMID:28649001; PMCID:PMC5517338.
Ater JL, Zhou T, Holmes E, Mazewski CM, Booth TN, Freyer DR, Lazarus KH, Packer RJ, Prados M, Sposto R, Vezina G, Wisoff JH, Pollack IF. Randomized study of two chemotherapy regimens for treatment of low-grade glioma in young children: a report from the Children’s Oncology Group. J Clin Oncol. 2012;30(21):2641-2647. doi:10.1200/jco.2011.36.6054. PMID:22665535; PMCID:PMC3413276.
Alvaro Lassaletta, Katrin Scheinemann, Shayna M. Zelcer, Juliette Hukin, Beverley A. Wilson, Nada Jabado, Anne Sophie Carret, Lucie Lafay-Cousin, et al. Phase II Weekly Vinblastine for Chemotherapy-Naïve Children With Progressive Low-Grade Glioma: A Canadian Pediatric Brain Tumor Consortium Study. JCO. 2016;34(29):3537-3543. doi:10.1200/jco.2016.68.1585.
Fangusaro J, Onar-Thomas A, Young Poussaint T, Wu S, Ligon AH, Lindeman N, Banerjee A, Packer RJ, Kilburn LB, Goldman S, Pollack IF, Qaddoumi I, Jakacki RI, Fisher PG, Dhall G, Baxter P, Kreissman SG, Stewart CF, Jones DTW, Pfister SM, Vezina G, Stern JS, Panigrahy A, Patay Z, Tamrazi B, Jones JY, Haque SS, Enterline DS, Cha S, Fisher MJ, Doyle LA, Smith M, Dunkel IJ, Fouladi M. Selumetinib in paediatric patients with BRAF-aberrant or neurofibromatosis type 1-associated recurrent, refractory, or progressive low-grade glioma: a multicentre, phase 2 trial. Lancet Oncol. 2019;20(7):1011-1022. doi:10.1016/s1470-2045(19)30277-3. PMID:31151904; PMCID:PMC6628202.
Anuradha Banerjee, Regina I. Jakacki, Arzu Onar-Thomas, Shengjie Wu, Theodore Nicolaides, Tina Young Poussaint, Jason Fangusaro, Joanna Phillips, et al. A phase I trial of the MEK inhibitor selumetinib (AZD6244) in pediatric patients with recurrent or refractory low-grade glioma: a Pediatric Brain Tumor Consortium (PBTC) study. Neuro-Oncology. 2017;19(8):1135-1144. doi:10.1093/neuonc/now282.
de Blank PMK, Orne-Ibekwe E, Packer R. International consensus recommendations for visual surveillance in optic pathway gliomas associated with neurofibromatosis type 1. J Neurooncol. 2020;148(3):571-578.
Listernick R, Louis DN, Packer RJ, Gutmann DH. Optic pathway gliomas in children with neurofibromatosis 1: consensus statement from the NF1 Optic Pathway Glioma Task Force. Ann Neurol. 1997;41(2):143-149. doi:10.1002/ana.410410204. PMID:9029062.
Copiez le texte de l'article et collez-le dans l'assistant IA de votre choix.
Article copié dans le presse-papiers
Ouvrez un assistant IA ci-dessous et collez le texte copié dans la conversation.