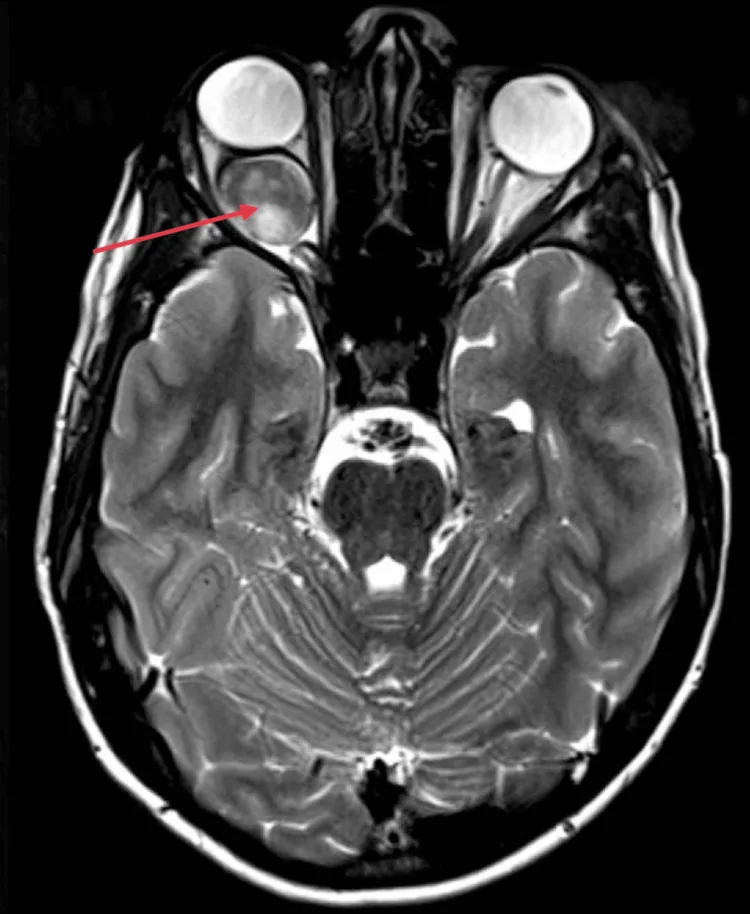
眼眶内视神经局限型
最常见的发生形式。主要症状为单眼视力下降和眼球突出。
局限于眼眶内的视神经,基本采取观察随访。 合并NF1的病例有自然缩小的报道。

视神经胶质瘤(optic nerve glioma / optic pathway glioma)是发生在视神经的一种胶质瘤。狭义上指发生于视交叉之前的视神经的胶质瘤。广义上指发生于包括视交叉后方在内的整个视路的胶质瘤(optic pathway glioma)。
组织学上,多数为良性的毛细胞型星形细胞瘤(pilocytic astrocytoma, WHO Grade I)。但也有部分恶性病例的报道。约70%发生于儿童期,是一种罕见的疾病,约占儿童脑肿瘤的0.5~5%。
与神经纤维瘤病1型(NF1, von Recklinghausen病)密切相关,约20~30%的视神经胶质瘤病例合并NF1。反之,NF1患者的眼眶病变中最常见的是视神经胶质瘤。
眼眶内视神经局限型
最常见的发生形式。主要症状为单眼视力下降和眼球突出。
局限于眼眶内的视神经,基本采取观察随访。 合并NF1的病例有自然缩小的报道。
视交叉浸润型
浸润至视交叉的类型。
导致双眼视力障碍,管理复杂。 多见于低龄发病,需评估向丘脑下部进展的情况。
视路-丘脑下部型
从视交叉后方进展至丘脑下部的类型。
可能合并内分泌异常(如生长障碍、性早熟等)。 治疗上需要神经外科与内分泌科协作。
根据遗传背景分类,包括NF1合并型(约占30%)和散发型(约占70%)。 NF1合并型可见双侧发生。
低龄儿童不会主动诉说视力下降。因此,家长或周围人常注意到斜视(尤其是内斜视),并首次就诊眼科。
无斜视的单眼视力下降更易延迟发现。初诊时可能已出现视神经萎缩。
双眼视神经胶质瘤多见于低龄发病者。常因视线异常或“看不见的行为”首次被察觉,视力障碍可能严重。
眼球突出不明显,也无疼痛。
视神经胶质瘤中,低龄儿童无法自觉或诉说视力下降,因此斜视(尤其是内斜视)常作为首发症状成为眼科就诊的契机。对于诊断为斜视的儿童,特别是单眼性斜视,应考虑视神经胶质瘤的可能性,并探讨包括视力、眼底及影像学检查在内的详细检查。
眼底检查常可见以下所见。
CT表现:
MRI表现(需进一步检查):
| 表现 | 标准 |
|---|---|
| 咖啡牛奶斑 | ≥6个(儿童:长径≥5mm,青春期后:长径≥15mm) |
| 神经纤维瘤 | ≥2个(任意类型)或1个及以上丛状神经纤维瘤 |
| 虹膜Lisch结节 | ≥2个 |
| 特征性骨病变 | 蝶骨翼发育不良或长骨皮质变薄 |
| 视神经胶质瘤 | 1个或以上 |
| 腋窝或腹股沟雀斑 | 存在 |
| 一级亲属 | 确诊为NF1者 |
确诊NF1需满足上述标准中的2项或以上。
MRI在评估肿瘤扩展范围方面优于CT,是详细检查所必需的。
| 表现 | 详细 |
|---|---|
| T1加权像 | 显示低信号 |
| Gd-DTPA增强效应 | 均匀肿大和明显增强效应 |
| 向下扭结 | NF1合并病例特征性(视神经向下弯曲) |
| 颅内扩展评估 | 确认视神经管内→颅内扩展、视交叉、下丘脑肿瘤的存在 |
视神经鞘脑膜瘤
最重要的鉴别疾病。
多见于成年女性,部分合并NF2。 CT/MRI上tram-track sign(轨道征)具有特征性,有助于与视神经胶质瘤鉴别。
视神经炎
常急性起病,多伴有眼球运动痛。
MRI显示视神经强化,但肿胀轻微。 类固醇治疗通常可改善。
其他鉴别诊断:
视神经胶质瘤表现为均匀增粗和向下弯曲,可与视神经鞘脑膜瘤的轨道征明确区分。
由于肿瘤为良性且多见于儿童,若局限于眶内视神经,原则上不进行手术切除或放射治疗。 基本方针是以定期影像学检查(MRI:每3~6个月)为中心的谨慎观察随访。
过去曾进行手术治疗,但由于导致不可逆失明的风险较高,目前倾向于避免手术切除。 合并NF1的病例有自然缩小的报道,因此尤其需要进行谨慎的观察随访。
当出现视力下降或肿瘤增大进展时,卡铂+长春新碱(CV方案)联合化疗被用作标准一线治疗3)4)。
CV方案的标准疗程(COG A9952等):
CV方案的客观缓解率(部分缓解+稳定)据报道为60~80%4)。
二线治疗选择:
对于化疗耐药的进展期病例可考虑放射治疗。但在儿童中,由于存在继发癌症风险、内分泌功能障碍(下丘脑附近照射)及认知功能影响等担忧,倾向于尽可能避免使用。
目前倾向于避免积极的手术切除。
考虑手术切除的情况:
视神经胶质瘤的组织病理学表现为良性的毛细胞型星形细胞瘤(pilocytic astrocytoma, WHO Grade I)。与胶质母细胞瘤(glioblastoma multiforme, WHO Grade IV)等高恶性度胶质瘤本质不同。
肿瘤细胞呈双极细胞突起的特征性形态,含有Rosenthal纤维。起源于视神经的胶质细胞(星形细胞),从内部压迫并取代视神经。
NF1(神经纤维瘤病1型)相关的视神经胶质瘤由**NF1基因(染色体17q11.2)**突变引起。
在散发性(不合并NF1)的毛细胞星形细胞瘤中,BRAF-KIAA1549融合基因常见。该融合基因也激活MAPK通路,促进肿瘤生长。
部分病例携带BRAF V600E突变,此类病例恶性程度往往较高6)。
大多数视神经胶质瘤为低级别,生长缓慢。肿瘤从内部使视神经肿大,在眼眶内导致视神经弯曲(kinking·downward kinking)。MRI上均匀肿大和向下弯曲是影像诊断的关键点。
局限于眼眶的病例:
视交叉-下丘脑浸润病例:
生命预后:
功能预后:
视力预后并非一致,进展型和稳定型病例并存,因此不仅需要评估肿瘤大小,还需进行视功能的纵向评估。即使MRI表现稳定,部分病例的视功能仍可能恶化;相反,合并NF1的病例有时会出现自然缩小。1, 8, 9)
已有报道显示MEK抑制剂对NF1相关低级别胶质瘤的有效性。
SPRINT试验(II期)报道了司美替尼对NF1相关进展性低级别胶质瘤(丛状神经纤维瘤)的客观缓解率为66%7)。目前正在探讨其应用于包括视神经胶质瘤在内的NF1相关低级别胶质瘤。
对于携带BRAF V600E突变的儿童低级别胶质瘤,达拉非尼+曲美替尼联合疗法已在临床试验中接受评估6)。 对于BRAF-KIAA1549融合阳性病例,BRAF抑制剂的疗效有限。
随着MEK抑制剂和BRAF抑制剂的问世,治疗正从传统化疗(CV方案)向基于分子谱的个体化治疗转变8)。 未来,基于基因突变谱(如BRAF融合、BRAF V600E、NF1突变等)的治疗选择可能会成为标准。
Listernick R, Charrow J, Greenwald M, Mets M. Natural history of optic pathway tumors in children with neurofibromatosis type 1: a longitudinal study. J Pediatr. 1994;125(1):63-66. doi:10.1016/s0022-3476(94)70122-9. PMID:8021787.
Ferner RE, Ferner RE, Huson SM, Thomas N, Moss C, Willshaw H, Evans DG, Upadhyaya M, Towers R, Gleeson M, Steiger C, Kirby A. Guidelines for the diagnosis and management of individuals with neurofibromatosis 1. J Med Genet. 2007;44(2):81-88. doi:10.1136/jmg.2006.045906. PMID:17105749; PMCID:PMC2598063.
Gnekow AK, Walker DA, Kandels D, Picton S, Giorgio Perilongo, Grill J, Stokland T, Sandstrom PE, Warmuth-Metz M, Pietsch T, Giangaspero F, Schmidt R, Faldum A, Kilmartin D, De Paoli A, De Salvo GL, of the Low Grade Glioma Consortium and the participating centers. A European randomised controlled trial of the addition of etoposide to standard vincristine and carboplatin induction as part of an 18-month treatment programme for childhood (≤16 years) low grade glioma - A final report. Eur J Cancer. 2017;81:206-225. doi:10.1016/j.ejca.2017.04.019. PMID:28649001; PMCID:PMC5517338.
Ater JL, Zhou T, Holmes E, Mazewski CM, Booth TN, Freyer DR, Lazarus KH, Packer RJ, Prados M, Sposto R, Vezina G, Wisoff JH, Pollack IF. Randomized study of two chemotherapy regimens for treatment of low-grade glioma in young children: a report from the Children’s Oncology Group. J Clin Oncol. 2012;30(21):2641-2647. doi:10.1200/jco.2011.36.6054. PMID:22665535; PMCID:PMC3413276.
Alvaro Lassaletta, Katrin Scheinemann, Shayna M. Zelcer, Juliette Hukin, Beverley A. Wilson, Nada Jabado, Anne Sophie Carret, Lucie Lafay-Cousin, et al. Phase II Weekly Vinblastine for Chemotherapy-Naïve Children With Progressive Low-Grade Glioma: A Canadian Pediatric Brain Tumor Consortium Study. JCO. 2016;34(29):3537-3543. doi:10.1200/jco.2016.68.1585.
Fangusaro J, Onar-Thomas A, Young Poussaint T, Wu S, Ligon AH, Lindeman N, Banerjee A, Packer RJ, Kilburn LB, Goldman S, Pollack IF, Qaddoumi I, Jakacki RI, Fisher PG, Dhall G, Baxter P, Kreissman SG, Stewart CF, Jones DTW, Pfister SM, Vezina G, Stern JS, Panigrahy A, Patay Z, Tamrazi B, Jones JY, Haque SS, Enterline DS, Cha S, Fisher MJ, Doyle LA, Smith M, Dunkel IJ, Fouladi M. Selumetinib in paediatric patients with BRAF-aberrant or neurofibromatosis type 1-associated recurrent, refractory, or progressive low-grade glioma: a multicentre, phase 2 trial. Lancet Oncol. 2019;20(7):1011-1022. doi:10.1016/s1470-2045(19)30277-3. PMID:31151904; PMCID:PMC6628202.
Anuradha Banerjee, Regina I. Jakacki, Arzu Onar-Thomas, Shengjie Wu, Theodore Nicolaides, Tina Young Poussaint, Jason Fangusaro, Joanna Phillips, et al. A phase I trial of the MEK inhibitor selumetinib (AZD6244) in pediatric patients with recurrent or refractory low-grade glioma: a Pediatric Brain Tumor Consortium (PBTC) study. Neuro-Oncology. 2017;19(8):1135-1144. doi:10.1093/neuonc/now282.
de Blank PMK, Orne-Ibekwe E, Packer R. International consensus recommendations for visual surveillance in optic pathway gliomas associated with neurofibromatosis type 1. J Neurooncol. 2020;148(3):571-578.
Listernick R, Louis DN, Packer RJ, Gutmann DH. Optic pathway gliomas in children with neurofibromatosis 1: consensus statement from the NF1 Optic Pathway Glioma Task Force. Ann Neurol. 1997;41(2):143-149. doi:10.1002/ana.410410204. PMID:9029062.