眼窩内視神経限局型
最多の発生形式。片眼性視力低下と眼球突出が主症状。
眼窩内の視神経に限局し、経過観察が基本となる。 NF1合併例では自然縮小の報告もある。

視神経膠腫(optic nerve glioma / optic pathway glioma)は、視神経に発生する膠腫(グリオーマ)の一種である。 狭義には視交叉より前の視神経に生じた膠腫を指す。 広義には視交叉より後方も含めた視路全体に生じた膠腫(optic pathway glioma)を意味する。
組織学的には、多くが良性の毛様細胞性星細胞腫(pilocytic astrocytoma, WHO Grade I)である。 ただし一部に悪性例の報告もある。 約70%が小児期発症であり、小児脳腫瘍の約0.5〜5%を占める稀な疾患である。
神経線維腫症1型(NF1, von Recklinghausen病)との関連が強く、視神経膠腫症例の約20〜30%でNF1の合併が認められる。 逆に、NF1患者の眼窩病変としては視神経膠腫が最も多い。
眼窩内視神経限局型
最多の発生形式。片眼性視力低下と眼球突出が主症状。
眼窩内の視神経に限局し、経過観察が基本となる。 NF1合併例では自然縮小の報告もある。
視交叉浸潤型
視交叉まで浸潤する型。
両眼性の視力障害を来たし、管理が複雑となる。 低年齢発症に多く、視床下部への進展を評価する。
視路・視床下部型
視交叉後方から視床下部まで進展する型。
内分泌異常(成長障害・思春期早発など)を合併することがある。 治療上、神経外科・内分泌科との連携が必要となる。
遺伝的背景による分類として、NF1合併型(全体の約30%)と孤発型(約70%)がある。 NF1合併型では両側性の発生もみられる。
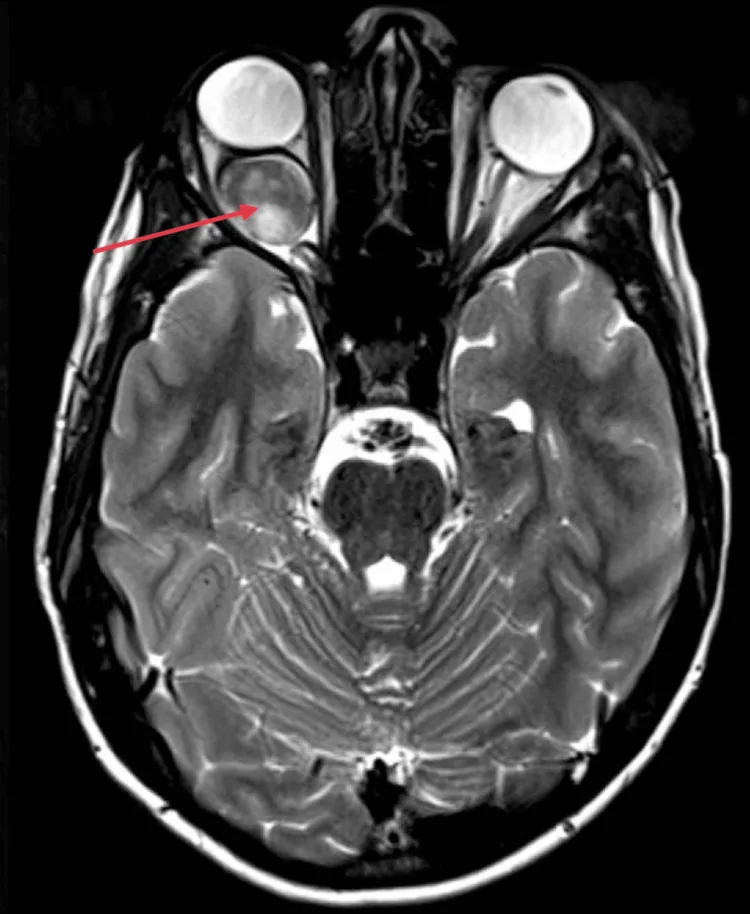
低年齢の小児は視力低下を自分から訴えない。 そのため、親や周囲の人が**斜視(特に内斜視)**に気づき、眼科を初診することが多い。
斜視のない片眼性視力低下はさらに発見が遅れやすい。 初診時にすでに視神経萎縮を来たしている場合もある。
両眼性の視神経膠腫は低年齢発症例に多い。 視線の異常や「見えていない行動」で初めて気づかれ、視力障害が重篤なことがある。
眼球突出は明らかでなく、痛みもない。
視神経膠腫では低年齢の小児が視力低下を自覚・訴えられないため、斜視(特に内斜視)が初発症状として眼科受診のきっかけとなることが多い。斜視と診断された小児、特に片眼性の斜視では視神経膠腫を念頭に、視力・眼底・画像検査を含めた精査を検討することが重要である。
眼底検査では以下の所見を認めることが多い。
NF1(神経線維腫症1型)合併例では、以下の全身所見を伴う。
CT所見:
MRI所見(精査の要):
NF1(神経線維腫症1型)の診断基準の主な項目を以下に示す2)。
| 所見 | 基準 |
|---|---|
| カフェオレ斑 | ≥6個(小児:長径≥5mm、思春期以降:長径≥15mm) |
| 神経線維腫 | ≥2個(任意の型)または1個以上の叢状神経線維腫 |
| 虹彩Lisch結節 | ≥2個 |
| 特徴的な骨病変 | 蝶形骨翼形成異常または長管骨皮質菲薄化 |
| 視神経膠腫 | 1個以上 |
| 腋窩・鼠径部の雀斑 | 存在する |
| 一親等内家族 | NF1の確定診断のある者 |
NF1の確定診断には上記基準の2項目以上を満たすことが必要である。
MRIはCTより腫瘍の進展範囲評価に優れ、精査に必須の検査である。
| 所見 | 詳細 |
|---|---|
| T1強調像 | 低信号を示す |
| Gd-DTPA造影効果 | 均一な腫大と強い造影効果を呈する |
| downward kinking | NF1合併例で特徴的(視神経の下方屈曲) |
| 頭蓋内進展評価 | 視神経管内→頭蓋内への進展・視交叉・視床下部の腫瘍の存在を確認 |
視神経鞘髄膜腫
鑑別の最重要疾患。
成人女性に多く、NF2合併例がある。 CT/MRIでtram-track sign(電車軌道様所見)が特徴的で、視神経膠腫との鑑別に有用。
視神経炎
急性発症・眼球運動痛を伴うことが多い。
MRIで視神経の造影増強を認めるが、腫大は軽度。 ステロイド治療で改善することが多い。
その他の鑑別疾患:
視神経膠腫は均一な腫大・downward kinkingを示し、視神経鞘髄膜腫のtram-track signとは明確に区別できる。
腫瘍は良性であり小児に多いため、眼窩内視神経に限局して存在する場合は外科切除・放射線照射を原則として行わない。 定期的な画像検査(MRI:3〜6ヶ月毎)を中心とした慎重な経過観察が基本方針である。
かつては手術が行われていたが、不可逆的な失明に至るリスクが高いため、現在では外科切除を避ける傾向にある。 NF1合併例では自然縮小(regression)の報告もあるため、特に慎重な経過観察が行われる。
視力低下・腫瘍増大が進行する場合には、カルボプラチン+ビンクリスチン(CV療法)の併用化学療法が標準的一次治療として用いられる3)4)。
CV療法の標準レジメン(COG A9952等):
CV療法の客観的奏効率(部分寛解+安定化)は60〜80%と報告されている4)。
二次治療の選択肢:
化学療法抵抗性の進行例に考慮される。 ただし、小児では二次がんリスク・内分泌機能障害(視床下部近傍照射)・認知機能への影響が懸念されるため、可能な限り避ける傾向にある。
現在は積極的な手術切除を避ける傾向にある。
適応が検討される状況:
視神経膠腫の病理組織学的所見は、良性の**毛様細胞性星細胞腫(pilocytic astrocytoma, WHO Grade I)**である。 膠芽腫(glioblastoma multiforme, WHO Grade IV)など高悪性度の膠腫とは本質的に異なる。
腫瘍細胞は双極性の細胞突起を持つ特徴的な形態を示し、Rosenthal線維を含む。 視神経のグリア細胞(アストロサイト)から発生し、視神経を内部から圧迫・置換する。
NF1(神経線維腫症1型)関連の視神経膠腫は、**NF1遺伝子(染色体17q11.2)**の変異に起因する。
孤発性(NF1非合併)の毛様細胞性星細胞腫では、BRAF-KIAA1549融合遺伝子が高頻度にみられる。 この融合遺伝子もMAPK経路を活性化させ、腫瘍増殖を促進する。
一部の症例ではBRAF V600E変異を持ち、これを有する症例は悪性度が高い傾向がある6)。
視神経膠腫の大多数は**低悪性度(low-grade)**であり、緩徐な増殖を示す。 腫瘍は視神経を内部から腫大させ、眼窩内で視神経を屈曲(kinking・downward kinking)させる。 MRIでの均一な腫大・下方屈曲が画像診断のキーポイントとなる。
眼窩内限局例:
視交叉・視床下部浸潤例:
生命予後:
機能予後:
視力予後は一様ではなく、進行例と安定例が混在するため、腫瘍サイズだけでなく視機能の縦断評価が必要である。MRI所見が安定していても視機能が悪化する症例があり、逆にNF1合併例では自然縮小を示すこともある。1, 8, 9)
NF1関連低悪性度膠腫に対するMEK阻害薬の有効性が報告されている。
SPRINT試験(Phase II)では、NF1関連進行性低悪性度膠腫(叢状神経線維腫)に対するセルメチニブの客観的奏効率66%が報告された7)。 視神経膠腫を含むNF1関連低悪性度膠腫への応用が検討されている。
BRAF V600E変異を持つ小児低悪性度膠腫に対し、ダブラフェニブ+トラメチニブの併用療法が臨床試験で評価されている6)。 BRAF-KIAA1549融合陽性例には、BRAF阻害薬の有効性は限定的である。
MEK阻害薬・BRAF阻害薬の登場により、従来の化学療法(CV療法)から分子プロファイルに基づく個別化治療へのシフトが進んでいる8)。 将来的には遺伝子変異プロファイル(BRAF融合、BRAF V600E、NF1変異等)に基づいた治療選択が標準化される可能性がある。
Listernick R, Charrow J, Greenwald M, Mets M. Natural history of optic pathway tumors in children with neurofibromatosis type 1: a longitudinal study. J Pediatr. 1994;125(1):63-66. doi:10.1016/s0022-3476(94)70122-9. PMID:8021787.
Ferner RE, Ferner RE, Huson SM, Thomas N, Moss C, Willshaw H, Evans DG, Upadhyaya M, Towers R, Gleeson M, Steiger C, Kirby A. Guidelines for the diagnosis and management of individuals with neurofibromatosis 1. J Med Genet. 2007;44(2):81-88. doi:10.1136/jmg.2006.045906. PMID:17105749; PMCID:PMC2598063.
Gnekow AK, Walker DA, Kandels D, Picton S, Giorgio Perilongo, Grill J, Stokland T, Sandstrom PE, Warmuth-Metz M, Pietsch T, Giangaspero F, Schmidt R, Faldum A, Kilmartin D, De Paoli A, De Salvo GL, of the Low Grade Glioma Consortium and the participating centers. A European randomised controlled trial of the addition of etoposide to standard vincristine and carboplatin induction as part of an 18-month treatment programme for childhood (≤16 years) low grade glioma - A final report. Eur J Cancer. 2017;81:206-225. doi:10.1016/j.ejca.2017.04.019. PMID:28649001; PMCID:PMC5517338.
Ater JL, Zhou T, Holmes E, Mazewski CM, Booth TN, Freyer DR, Lazarus KH, Packer RJ, Prados M, Sposto R, Vezina G, Wisoff JH, Pollack IF. Randomized study of two chemotherapy regimens for treatment of low-grade glioma in young children: a report from the Children’s Oncology Group. J Clin Oncol. 2012;30(21):2641-2647. doi:10.1200/jco.2011.36.6054. PMID:22665535; PMCID:PMC3413276.
Alvaro Lassaletta, Katrin Scheinemann, Shayna M. Zelcer, Juliette Hukin, Beverley A. Wilson, Nada Jabado, Anne Sophie Carret, Lucie Lafay-Cousin, et al. Phase II Weekly Vinblastine for Chemotherapy-Naïve Children With Progressive Low-Grade Glioma: A Canadian Pediatric Brain Tumor Consortium Study. JCO. 2016;34(29):3537-3543. doi:10.1200/jco.2016.68.1585.
Fangusaro J, Onar-Thomas A, Young Poussaint T, Wu S, Ligon AH, Lindeman N, Banerjee A, Packer RJ, Kilburn LB, Goldman S, Pollack IF, Qaddoumi I, Jakacki RI, Fisher PG, Dhall G, Baxter P, Kreissman SG, Stewart CF, Jones DTW, Pfister SM, Vezina G, Stern JS, Panigrahy A, Patay Z, Tamrazi B, Jones JY, Haque SS, Enterline DS, Cha S, Fisher MJ, Doyle LA, Smith M, Dunkel IJ, Fouladi M. Selumetinib in paediatric patients with BRAF-aberrant or neurofibromatosis type 1-associated recurrent, refractory, or progressive low-grade glioma: a multicentre, phase 2 trial. Lancet Oncol. 2019;20(7):1011-1022. doi:10.1016/s1470-2045(19)30277-3. PMID:31151904; PMCID:PMC6628202.
Anuradha Banerjee, Regina I. Jakacki, Arzu Onar-Thomas, Shengjie Wu, Theodore Nicolaides, Tina Young Poussaint, Jason Fangusaro, Joanna Phillips, et al. A phase I trial of the MEK inhibitor selumetinib (AZD6244) in pediatric patients with recurrent or refractory low-grade glioma: a Pediatric Brain Tumor Consortium (PBTC) study. Neuro-Oncology. 2017;19(8):1135-1144. doi:10.1093/neuonc/now282.
de Blank PMK, Orne-Ibekwe E, Packer R. International consensus recommendations for visual surveillance in optic pathway gliomas associated with neurofibromatosis type 1. J Neurooncol. 2020;148(3):571-578.
Listernick R, Louis DN, Packer RJ, Gutmann DH. Optic pathway gliomas in children with neurofibromatosis 1: consensus statement from the NF1 Optic Pathway Glioma Task Force. Ann Neurol. 1997;41(2):143-149. doi:10.1002/ana.410410204. PMID:9029062.