Il glioma del nervo ottico (optic nerve glioma / optic pathway glioma) è un tipo di glioma che si sviluppa nel nervo ottico. In senso stretto, si riferisce a un glioma che si verifica nel nervo ottico prima del chiasma ottico. In senso lato, indica un glioma che si verifica in tutta la via ottica, inclusa la parte posteriore al chiasma (optic pathway glioma).
Istologicamente, la maggior parte sono astrocitomi pilocitici benigni (pilocytic astrocytoma, WHO Grade I). Tuttavia, sono stati riportati alcuni casi maligni. Circa il 70% si manifesta nell’infanzia ed è una malattia rara che rappresenta circa lo 0,5-5% dei tumori cerebrali pediatrici.
Esiste una forte associazione con la neurofibromatosi di tipo 1 (NF1, malattia di von Recklinghausen), e circa il 20-30% dei casi di glioma del nervo ottico presenta NF1. Al contrario, il glioma del nervo ottico è la lesione orbitale più comune nei pazienti con NF1.
Classificazione in base alla sede e al background genetico
Forma di insorgenza più frequente. I sintomi principali sono perdita della vista monolaterale e protrusione del bulbo oculare.
Limitato al nervo ottico all’interno dell’orbita, il trattamento di base è l’osservazione. Nei casi associati a NF1 sono state riportate regressioni spontanee.
Tipo infiltrante il chiasma ottico
Tipo che infiltra fino al chiasma ottico.
Causa disturbi visivi bilaterali e la gestione è complessa. Si manifesta spesso in età precoce; valutare l’estensione all’ipotalamo.
Tipo ottico-ipotalamico
Tipo che si estende dal chiasma ottico posteriore all’ipotalamo.
Può associarsi ad anomalie endocrine (disturbi della crescita, pubertà precoce, ecc.). Il trattamento richiede la collaborazione tra neurochirurgia ed endocrinologia.
La classificazione in base al background genetico comprende il tipo associato a NF1 (circa il 30% dei casi) e il tipo sporadico (circa il 70%). Nel tipo associato a NF1 si osserva anche una comparsa bilaterale.
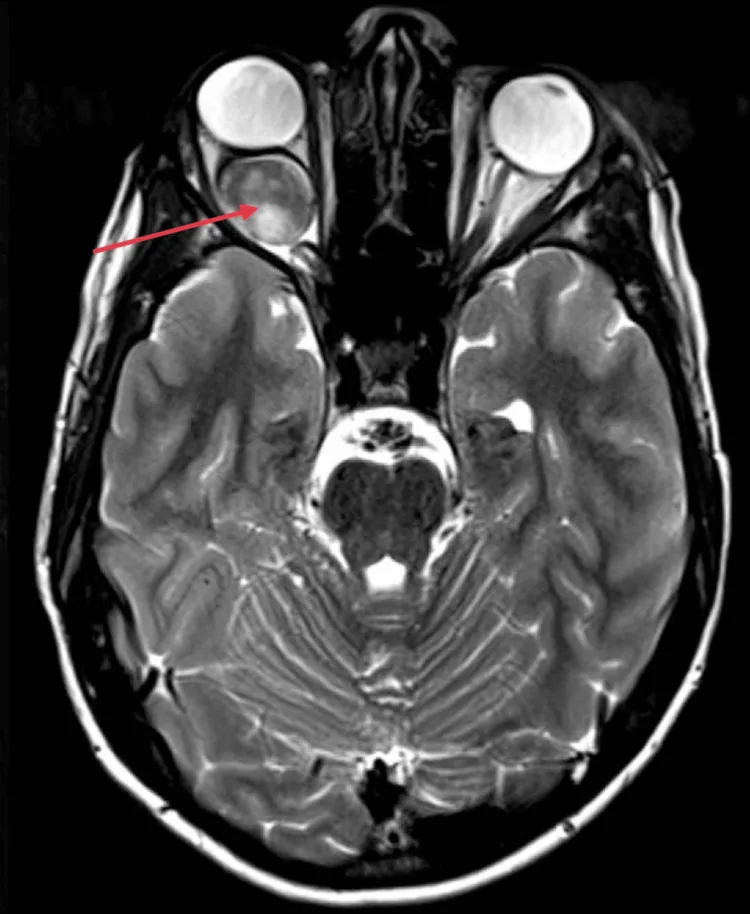
Kotecha MR, et al. Idiopathic Optic Nerve Glioma: A Case Report. Cureus. 2024. Figure 2. PMCID: PMC11744336. License: CC BY.
Nella MRI assiale, il nervo ottico nell’orbita destra appare fusiformemente ingrossato, con un potenziamento omogeneo e intenso (freccia rossa), e si osserva proptosi dovuta all’effetto massa sulla parte posteriore del bulbo oculare. Corrisponde ai reperti MRI (ingrossamento omogeneo del nervo ottico, downward kinking) trattati nella sezione «2. Principali sintomi e reperti clinici».
I bambini piccoli non riferiscono spontaneamente una riduzione della vista. Per questo motivo, spesso sono i genitori o le persone intorno a notare strabismo (in particolare esotropia) e portare il bambino a una prima visita oculistica.
La riduzione della vista monolaterale senza strabismo è ancora più facile da scoprire tardivamente. A volte alla prima visita è già presente atrofia del nervo ottico.
Il glioma del nervo ottico bilaterale è più comune nei casi a insorgenza precoce. Viene notato per la prima volta a causa di anomalie dello sguardo o di “comportamenti che indicano mancata visione”, e il deficit visivo può essere grave.
L’esoftalmo non è evidente e non c’è dolore.
QSe a un bambino viene diagnosticato uno strabismo, è possibile che si tratti di un glioma del nervo ottico?
A
Nel glioma del nervo ottico, i bambini piccoli spesso non riescono a percepire o riferire una riduzione della vista, quindi lo strabismo (in particolare l’esotropia) è spesso il sintomo iniziale che porta a una visita oculistica. Nei bambini con diagnosi di strabismo, specialmente se monolaterale, è importante considerare la possibilità di un glioma del nervo ottico e valutare un approfondimento che includa esami della vista, del fondo oculare e di imaging.
Nelle lesioni orbitali da NF1, il glioma ottico è il più comune
QSe si ha NF1 (neurofibromatosi di tipo 1), si è più inclini a sviluppare un glioma del nervo ottico?
A
I pazienti con NF1 hanno un rischio significativamente più elevato di glioma del nervo ottico, e il 20-30% di tutti i casi di glioma del nervo ottico sono associati a NF11). Dopo la diagnosi di NF1, si raccomanda un follow-up oftalmologico regolare come screening per il glioma del nervo ottico. Al contrario, quando viene scoperto un glioma del nervo ottico in un bambino, è importante verificare in tutti i casi se vengono soddisfatti i criteri diagnostici per NF1.
Malattia più importante nella diagnosi differenziale.
Più comune nelle donne adulte, con possibile associazione a NF2.
Alla TC/RM è caratteristico il tram-track sign (segno del binario), utile per la differenziazione dal glioma del nervo ottico.
Neurite ottica
Spesso si manifesta con esordio acuto e dolore al movimento oculare.
Alla RMN si osserva un potenziamento del contrasto del nervo ottico, ma il rigonfiamento è lieve. Spesso migliora con la terapia steroidea.
Il glioma del nervo ottico mostra un ingrossamento uniforme e un downward kinking, chiaramente distinguibile dal tram-track sign del meningioma della guaina del nervo ottico.
QQual è la differenza tra glioma del nervo ottico e meningioma della guaina del nervo ottico?
A
Il glioma del nervo ottico è un tumore benigno (astrocitoma pilocitico) comune nei bambini, spesso associato a NF1. Alla TC/RM mostra un ingrossamento uniforme del nervo ottico e un downward kinking caratteristici. Il meningioma della guaina del nervo ottico è più frequente nelle donne adulte, talvolta associato a NF2, e si differenzia per la presenza del tram-track sign (segno a binario ferroviario: calcificazione o enhancement lungo la guaina del nervo ottico) alla TC/RM.
Poiché il tumore è benigno e comune nei bambini, se è limitato al nervo ottico intraorbitario, di norma non si esegue resezione chirurgica né radioterapia. La strategia di base è un’attenta osservazione clinica con controlli periodici di imaging (RM ogni 3-6 mesi).
In passato si ricorreva all’intervento chirurgico, ma a causa dell’alto rischio di cecità irreversibile, oggi si tende a evitare la resezione chirurgica. Nei casi associati a NF1 sono state riportate regressioni spontanee, pertanto si effettua un’osservazione particolarmente attenta.
Chemioterapia (casi progressivi o con calo visivo)
In caso di progressione del calo visivo o dell’aumento del tumore, la chemioterapia combinata con carboplatino e vincristina (terapia CV) viene utilizzata come trattamento standard di prima linea3)4).
Regime standard di CV (COG A9952, ecc.):
Vincristina: 1,5 mg/m² ev, 1 volta a settimana × 10 settimane
Carboplatino: 550 mg/m² ev, ogni 3 settimane
Il tasso di risposta obiettiva (remissione parziale + stabilizzazione) della terapia CV è riportato essere del 60-80%4).
Opzioni di trattamento di seconda linea:
Cisplatino + etoposide
Temozolomide (agente alchilante)
Vinblastina in monoterapia (recidiva dopo terapia CV 5))
La radioterapia è considerata nei casi avanzati resistenti alla chemioterapia. Tuttavia, nei bambini si tende a evitarla il più possibile a causa del rischio di tumori secondari, disfunzioni endocrine (irradiazione vicino all’ipotalamo) e impatto sulle funzioni cognitive.
L’esame istopatologico del glioma del nervo ottico mostra un astrocitoma pilocitico (pilocytic astrocytoma, WHO Grade I) benigno. È essenzialmente diverso dai gliomi ad alto grado come il glioblastoma multiforme (WHO Grade IV).
Le cellule tumorali mostrano una morfologia caratteristica con prolungamenti cellulari bipolari e contengono fibre di Rosenthal. Originano dalle cellule gliali (astrociti) del nervo ottico, comprimendo e sostituendo il nervo dall’interno.
Il glioma del nervo ottico associato a NF1 (neurofibromatosi di tipo 1) è causato da mutazioni del gene NF1 (cromosoma 17q11.2).
Il gene NF1 è un gene oncosoppressore che codifica per la proteina neurofibromina.
neurofibromina funge da proteina attivatrice della Ras-GTPasi (GAP) e sopprime il segnale di Ras
Mutazione di NF1 → perdita di funzione della neurofibromina → attivazione costitutiva della via del segnale di Ras → potenziamento del segnale MAPK → proliferazione incontrollata delle cellule gliali
Negli astrocitomi pilocitici sporadici (non associati a NF1), si riscontra frequentemente il gene di fusione BRAF-KIAA1549. Anche questo gene di fusione attiva la via MAPK, promuovendo la crescita tumorale.
Alcuni casi presentano la mutazione BRAF V600E, e questi casi tendono ad avere un grado di malignità più elevato6).
La maggior parte dei gliomi del nervo ottico sono di basso grado (low-grade) e mostrano una crescita lenta.
Il tumore ingrossa il nervo ottico dall’interno e provoca una flessione (kinking o downward kinking) del nervo all’interno dell’orbita.
L’ingrossamento uniforme e la flessione verso il basso alla risonanza magnetica sono punti chiave per la diagnosi per immagini.
La prognosi visiva non è uniforme: casi progressivi e stabili coesistono, pertanto è necessaria una valutazione longitudinale non solo delle dimensioni tumorali ma anche della funzione visiva. In alcuni casi la funzione visiva peggiora nonostante reperti MRI stabili, mentre nei pazienti con NF1 si può osservare una regressione spontanea. 1, 8, 9)
Valutazione della funzione visiva: misurare ripetutamente acuità visiva, campo visivo, visione dei colori e RAPD in base all’età
Follow-up per immagini: monitorare la ricrescita, l’estensione al chiasma e l’estensione intracranica con MRI8)
Valutazione endocrina: nei casi con estensione ipotalamica, verificare disturbi della crescita, pubertà precoce e diabete insipido
Gestione sistemica della NF1: eseguire un follow-up sistemico parallelo che includa lesioni cutanee, altri tumori e aspetti dello sviluppo2, 9)
È stata riportata l’efficacia degli inibitori di MEK per i gliomi di basso grado associati a NF1.
Nello studio SPRINT (Fase II), è stato riportato un tasso di risposta obiettiva del 66% con selumetinib per i gliomi di basso grado progressivi associati a NF1 (neurofibromi plessiformi) 7).
Si sta valutando l’applicazione ai gliomi di basso grado associati a NF1, inclusi i gliomi del nervo ottico.
Per i gliomi di basso grado pediatrici con mutazione BRAF V600E, la terapia combinata con dabrafenib + trametinib è stata valutata in studi clinici6).
Nei casi con fusione BRAF-KIAA1549 positiva, l’efficacia degli inibitori di BRAF è limitata.
Con l’introduzione degli inibitori di MEK e BRAF, si sta passando dalla chemioterapia convenzionale (terapia CV) a trattamenti personalizzati basati sul profilo molecolare8).
In futuro, la scelta terapeutica potrebbe essere standardizzata in base al profilo di mutazione genetica (fusione BRAF, BRAF V600E, mutazione NF1, ecc.).
Listernick R, Charrow J, Greenwald M, Mets M. Natural history of optic pathway tumors in children with neurofibromatosis type 1: a longitudinal study. J Pediatr. 1994;125(1):63-66. doi:10.1016/s0022-3476(94)70122-9. PMID:8021787.
Ferner RE, Ferner RE, Huson SM, Thomas N, Moss C, Willshaw H, Evans DG, Upadhyaya M, Towers R, Gleeson M, Steiger C, Kirby A. Guidelines for the diagnosis and management of individuals with neurofibromatosis 1. J Med Genet. 2007;44(2):81-88. doi:10.1136/jmg.2006.045906. PMID:17105749; PMCID:PMC2598063.
Gnekow AK, Walker DA, Kandels D, Picton S, Giorgio Perilongo, Grill J, Stokland T, Sandstrom PE, Warmuth-Metz M, Pietsch T, Giangaspero F, Schmidt R, Faldum A, Kilmartin D, De Paoli A, De Salvo GL, of the Low Grade Glioma Consortium and the participating centers. A European randomised controlled trial of the addition of etoposide to standard vincristine and carboplatin induction as part of an 18-month treatment programme for childhood (≤16 years) low grade glioma - A final report. Eur J Cancer. 2017;81:206-225. doi:10.1016/j.ejca.2017.04.019. PMID:28649001; PMCID:PMC5517338.
Ater JL, Zhou T, Holmes E, Mazewski CM, Booth TN, Freyer DR, Lazarus KH, Packer RJ, Prados M, Sposto R, Vezina G, Wisoff JH, Pollack IF. Randomized study of two chemotherapy regimens for treatment of low-grade glioma in young children: a report from the Children’s Oncology Group. J Clin Oncol. 2012;30(21):2641-2647. doi:10.1200/jco.2011.36.6054. PMID:22665535; PMCID:PMC3413276.
Alvaro Lassaletta, Katrin Scheinemann, Shayna M. Zelcer, Juliette Hukin, Beverley A. Wilson, Nada Jabado, Anne Sophie Carret, Lucie Lafay-Cousin, et al. Phase II Weekly Vinblastine for Chemotherapy-Naïve Children With Progressive Low-Grade Glioma: A Canadian Pediatric Brain Tumor Consortium Study. JCO. 2016;34(29):3537-3543. doi:10.1200/jco.2016.68.1585.
Fangusaro J, Onar-Thomas A, Young Poussaint T, Wu S, Ligon AH, Lindeman N, Banerjee A, Packer RJ, Kilburn LB, Goldman S, Pollack IF, Qaddoumi I, Jakacki RI, Fisher PG, Dhall G, Baxter P, Kreissman SG, Stewart CF, Jones DTW, Pfister SM, Vezina G, Stern JS, Panigrahy A, Patay Z, Tamrazi B, Jones JY, Haque SS, Enterline DS, Cha S, Fisher MJ, Doyle LA, Smith M, Dunkel IJ, Fouladi M. Selumetinib in paediatric patients with BRAF-aberrant or neurofibromatosis type 1-associated recurrent, refractory, or progressive low-grade glioma: a multicentre, phase 2 trial. Lancet Oncol. 2019;20(7):1011-1022. doi:10.1016/s1470-2045(19)30277-3. PMID:31151904; PMCID:PMC6628202.
Anuradha Banerjee, Regina I. Jakacki, Arzu Onar-Thomas, Shengjie Wu, Theodore Nicolaides, Tina Young Poussaint, Jason Fangusaro, Joanna Phillips, et al. A phase I trial of the MEK inhibitor selumetinib (AZD6244) in pediatric patients with recurrent or refractory low-grade glioma: a Pediatric Brain Tumor Consortium (PBTC) study. Neuro-Oncology. 2017;19(8):1135-1144. doi:10.1093/neuonc/now282.
de Blank PMK, Orne-Ibekwe E, Packer R. International consensus recommendations for visual surveillance in optic pathway gliomas associated with neurofibromatosis type 1. J Neurooncol. 2020;148(3):571-578.
Listernick R, Louis DN, Packer RJ, Gutmann DH. Optic pathway gliomas in children with neurofibromatosis 1: consensus statement from the NF1 Optic Pathway Glioma Task Force. Ann Neurol. 1997;41(2):143-149. doi:10.1002/ana.410410204. PMID:9029062.
Copia il testo dell'articolo e incollalo nell'assistente IA che preferisci.
Articolo copiato negli appunti
Apri un assistente IA qui sotto e incolla il testo copiato nella chat.