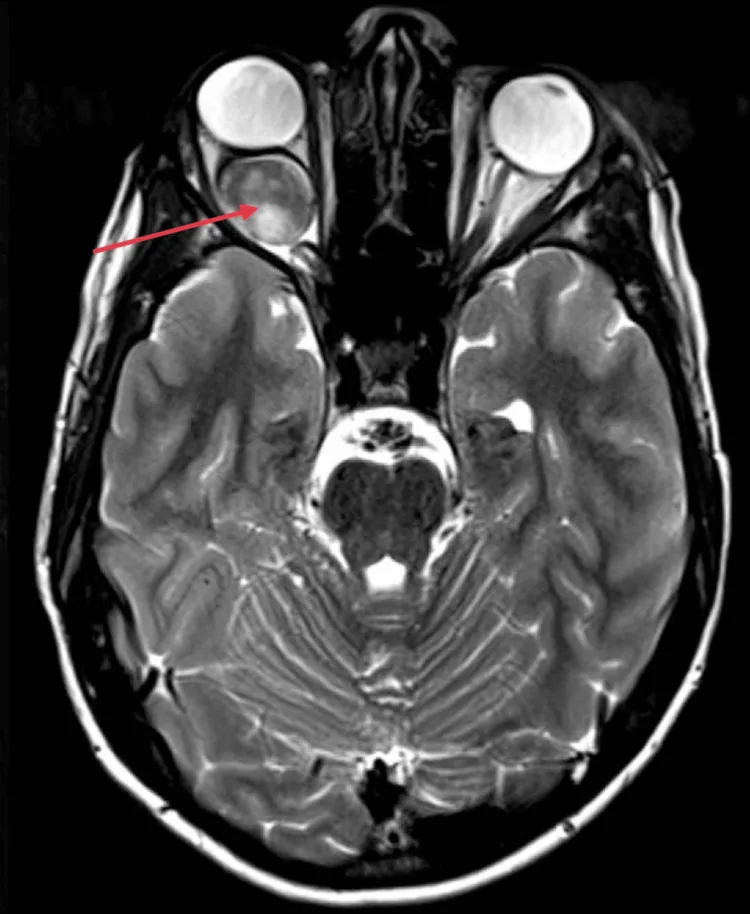
眼眶內視神經局限型
最常見的發生形式。主要症狀為單眼視力下降和眼球突出。
局限於眼眶內的視神經,基本以觀察追蹤為主。 合併NF1的病例有自然縮小的報告。

視神經膠質瘤(optic nerve glioma / optic pathway glioma)是發生在視神經的一種神經膠質瘤。狹義上指發生在視交叉之前的視神經的膠質瘤。廣義上指發生在視交叉之後的整個視路的神經膠質瘤(optic pathway glioma)。
組織學上,多數為良性的毛細胞型星狀細胞瘤(pilocytic astrocytoma, WHO Grade I)。但部分也有惡性病例的報告。約70%發生於兒童期,是佔兒童腦腫瘤約0.5~5%的罕見疾病。
與神經纖維瘤病第1型(NF1, von Recklinghausen病)有強烈關聯,視神經膠質瘤病例中約20~30%合併有NF1。反之,NF1患者的眼眶病變中最常見的是視神經膠質瘤。
眼眶內視神經局限型
最常見的發生形式。主要症狀為單眼視力下降和眼球突出。
局限於眼眶內的視神經,基本以觀察追蹤為主。 合併NF1的病例有自然縮小的報告。
視交叉浸潤型
浸潤至視交叉的類型。
會造成雙眼視力障礙,管理上較為複雜。 多發生於低年齡層,需評估是否向視丘下部進展。
視路・視丘下部型
從視交叉後方進展至視丘下部的類型。
可能合併內分泌異常(生長障礙、性早熟等)。 治療上需與神經外科、內分泌科合作。
依遺傳背景分類,可分為NF1合併型(約占30%)與孤發型(約占70%)。 NF1合併型可能出現雙側發生。
年幼兒童不會主動抱怨視力下降。因此,家長或周圍的人常會注意到斜視(特別是內斜視),並首次帶孩子到眼科就診。
無斜視的單眼視力下降更難被發現。初診時可能已出現視神經萎縮。
雙側視神經膠質瘤多見於低齡發病者。常因視線異常或「看不見的行為」才被發現,視力障礙可能已很嚴重。
眼球突出不明顯,也無疼痛。
視神經膠質瘤患者中,年幼兒童常因無法自覺或表達視力下降,而以斜視(特別是內斜視)作為初發症狀,成為就診契機。對於被診斷為斜視的兒童,尤其是單眼性斜視,應考慮視神經膠質瘤的可能性,並進行包括視力、眼底及影像檢查在內的詳細評估。
眼底檢查常可發現以下所見。
NF1(神經纖維瘤病第1型)合併案例中,伴隨以下全身性所見。
CT表現:
MRI所見(需詳細檢查):
| 表現 | 標準 |
|---|---|
| 咖啡牛奶斑 | ≥6個(兒童:長徑≥5mm,青春期後:長徑≥15mm) |
| 神經纖維瘤 | 2個以上(任何類型)或1個以上叢狀神經纖維瘤 |
| 虹膜Lisch結節 | 2個以上 |
| 特徵性骨骼病變 | 蝶骨翼發育異常或長骨皮質變薄 |
| 視神經膠質瘤 | 1個以上 |
| 腋窩或腹股溝雀斑 | 存在 |
| 一等親內家族 | 確診為NF1者 |
確診NF1需滿足上述標準中的2項或以上。
MRI比CT更擅長評估腫瘤的擴展範圍,是詳細檢查的必要檢查。
| 所見 | 詳細 |
|---|---|
| T1加權像 | 呈現低信號 |
| Gd-DTPA對比增強效果 | 呈現均勻腫大與強烈對比增強 |
| 向下扭結(downward kinking) | NF1合併病例特徵(視神經向下彎曲) |
| 顱內進展評估 | 確認視神經管→顱內進展、視交叉、下視丘腫瘤的存在 |
視神經鞘腦膜瘤
最重要的鑑別疾病。
多見於成年女性,部分合併NF2。 CT/MRI上tram-track sign(電車軌道樣徵象)具有特徵性,有助於與視神經膠質瘤鑑別。
視神經炎
常為急性發作,且多伴有眼球運動痛。
MRI顯示視神經有顯影增強,但腫大程度輕微。 類固醇治療通常可改善。
其他鑑別診斷:
視神經膠質瘤呈現均勻腫大及向下彎曲,可與視神經鞘腦膜瘤的軌道徵明確區分。
腫瘤為良性且多見於兒童,若僅局限於眼眶內視神經,原則上不進行手術切除或放射治療。基本方針是以定期影像檢查(MRI:每3~6個月)為主的謹慎觀察追蹤。
過去曾採用手術治療,但因導致不可逆失明的風險較高,目前傾向避免手術切除。合併NF1的病例有自然縮小的報告,因此會進行更謹慎的觀察追蹤。
若出現視力下降或腫瘤增大進展,卡鉑+長春新鹼(CV療法)的聯合化療被用作標準一線治療3)4)。
CV療法標準方案(COG A9952等):
CV療法的客觀緩解率(部分緩解+穩定)報告為60〜80%4)。
二線治療選擇:
化學治療抵抗性的進展病例可考慮使用。 然而,在兒童中,由於擔心二次癌症風險、內分泌功能障礙(下視丘附近照射)以及對認知功能的影響,傾向於盡可能避免使用。
目前傾向避免積極的手術切除。
考慮手術的適應情況:
視神經膠瘤的病理組織學所見為良性的毛細胞型星狀細胞瘤(pilocytic astrocytoma, WHO Grade I)。 與膠質母細胞瘤(glioblastoma multiforme, WHO Grade IV)等高惡性度膠瘤本質上不同。
腫瘤細胞呈現雙極性細胞突起的特徵性形態,並含有Rosenthal纖維。 源自視神經的膠質細胞(星狀細胞),從內部壓迫並取代視神經。
NF1(神經纖維瘤病第1型)相關的視神經膠瘤,起因於**NF1基因(染色體17q11.2)**的突變。
在散發性(非 NF1 合併)的毛細胞星狀細胞瘤中,BRAF-KIAA1549 融合基因 常見。此融合基因也會活化 MAPK 路徑,促進腫瘤增生。
部分病例帶有 BRAF V600E 突變,有此突變的病例惡性度傾向較高6)。
大多數視神經膠質瘤屬於低度惡性(low-grade),生長緩慢。 腫瘤會從內部使視神經腫大,並在眼眶內造成視神經彎曲(kinking・downward kinking)。 MRI上均勻腫大及向下彎曲是影像診斷的關鍵。
侷限於眼眶內病例:
視交叉及下視丘浸潤案例:
生命預後:
功能預後:
視力預後並非一致,進展型與穩定型病例混合存在,因此不僅需要評估腫瘤大小,還需要進行視功能的縱向評估。即使MRI表現穩定,部分病例視功能仍會惡化;反之,NF1合併病例有時會出現自然縮小。1, 8, 9)
已有報告指出MEK抑制劑對NF1相關低度惡性膠質瘤具有療效。
SPRINT試驗(第二期)報告指出,司美替尼對NF1相關進展性低度惡性膠質瘤(叢狀神經纖維瘤)的客觀緩解率為66%7)。目前正在探討其應用於視神經膠質瘤等NF1相關低度惡性膠質瘤的可能性。
對於具有BRAF V600E突變的兒童低度惡性膠質瘤,臨床試驗正在評估達拉非尼+曲美替尼的聯合療法6)。 對於BRAF-KIAA1549融合陽性的病例,BRAF抑制劑的療效有限。
隨著MEK抑制劑和BRAF抑制劑的出現,治療正從傳統化療(CV療法)轉向基於分子特徵的個人化治療8)。 未來,根據基因突變特徵(如BRAF融合、BRAF V600E、NF1突變等)選擇治療方案可能成為標準。
Listernick R, Charrow J, Greenwald M, Mets M. Natural history of optic pathway tumors in children with neurofibromatosis type 1: a longitudinal study. J Pediatr. 1994;125(1):63-66. doi:10.1016/s0022-3476(94)70122-9. PMID:8021787.
Ferner RE, Ferner RE, Huson SM, Thomas N, Moss C, Willshaw H, Evans DG, Upadhyaya M, Towers R, Gleeson M, Steiger C, Kirby A. Guidelines for the diagnosis and management of individuals with neurofibromatosis 1. J Med Genet. 2007;44(2):81-88. doi:10.1136/jmg.2006.045906. PMID:17105749; PMCID:PMC2598063.
Gnekow AK, Walker DA, Kandels D, Picton S, Giorgio Perilongo, Grill J, Stokland T, Sandstrom PE, Warmuth-Metz M, Pietsch T, Giangaspero F, Schmidt R, Faldum A, Kilmartin D, De Paoli A, De Salvo GL, of the Low Grade Glioma Consortium and the participating centers. A European randomised controlled trial of the addition of etoposide to standard vincristine and carboplatin induction as part of an 18-month treatment programme for childhood (≤16 years) low grade glioma - A final report. Eur J Cancer. 2017;81:206-225. doi:10.1016/j.ejca.2017.04.019. PMID:28649001; PMCID:PMC5517338.
Ater JL, Zhou T, Holmes E, Mazewski CM, Booth TN, Freyer DR, Lazarus KH, Packer RJ, Prados M, Sposto R, Vezina G, Wisoff JH, Pollack IF. Randomized study of two chemotherapy regimens for treatment of low-grade glioma in young children: a report from the Children’s Oncology Group. J Clin Oncol. 2012;30(21):2641-2647. doi:10.1200/jco.2011.36.6054. PMID:22665535; PMCID:PMC3413276.
Alvaro Lassaletta, Katrin Scheinemann, Shayna M. Zelcer, Juliette Hukin, Beverley A. Wilson, Nada Jabado, Anne Sophie Carret, Lucie Lafay-Cousin, et al. Phase II Weekly Vinblastine for Chemotherapy-Naïve Children With Progressive Low-Grade Glioma: A Canadian Pediatric Brain Tumor Consortium Study. JCO. 2016;34(29):3537-3543. doi:10.1200/jco.2016.68.1585.
Fangusaro J, Onar-Thomas A, Young Poussaint T, Wu S, Ligon AH, Lindeman N, Banerjee A, Packer RJ, Kilburn LB, Goldman S, Pollack IF, Qaddoumi I, Jakacki RI, Fisher PG, Dhall G, Baxter P, Kreissman SG, Stewart CF, Jones DTW, Pfister SM, Vezina G, Stern JS, Panigrahy A, Patay Z, Tamrazi B, Jones JY, Haque SS, Enterline DS, Cha S, Fisher MJ, Doyle LA, Smith M, Dunkel IJ, Fouladi M. Selumetinib in paediatric patients with BRAF-aberrant or neurofibromatosis type 1-associated recurrent, refractory, or progressive low-grade glioma: a multicentre, phase 2 trial. Lancet Oncol. 2019;20(7):1011-1022. doi:10.1016/s1470-2045(19)30277-3. PMID:31151904; PMCID:PMC6628202.
Anuradha Banerjee, Regina I. Jakacki, Arzu Onar-Thomas, Shengjie Wu, Theodore Nicolaides, Tina Young Poussaint, Jason Fangusaro, Joanna Phillips, et al. A phase I trial of the MEK inhibitor selumetinib (AZD6244) in pediatric patients with recurrent or refractory low-grade glioma: a Pediatric Brain Tumor Consortium (PBTC) study. Neuro-Oncology. 2017;19(8):1135-1144. doi:10.1093/neuonc/now282.
de Blank PMK, Orne-Ibekwe E, Packer R. International consensus recommendations for visual surveillance in optic pathway gliomas associated with neurofibromatosis type 1. J Neurooncol. 2020;148(3):571-578.
Listernick R, Louis DN, Packer RJ, Gutmann DH. Optic pathway gliomas in children with neurofibromatosis 1: consensus statement from the NF1 Optic Pathway Glioma Task Force. Ann Neurol. 1997;41(2):143-149. doi:10.1002/ana.410410204. PMID:9029062.