Le rhabdomyosarcome est une tumeur maligne dérivée des cellules mésenchymateuses de l’orbite, montrant une différenciation musculaire striée. Il provient de cellules mésenchymateuses indifférenciées de l’orbite et non de la transformation maligne de tissu musculaire différencié comme les muscles extra-oculaires. C’est la tumeur maligne orbitaire la plus fréquente chez l’enfant ; devant une tumeur orbitaire à croissance rapide chez un enfant, il faut d’abord envisager ce diagnostic.
Aux États-Unis, il représente environ 5 % de tous les cancers pédiatriques, avec environ 250 nouveaux cas par an. Environ 10 % (environ 35 cas par an) sont d’origine orbitaire. L’incidence annuelle est estimée à 4 cas par million d’habitants dans la population générale et à 4,5 cas par million d’enfants1).
L’âge moyen au diagnostic est de 7 à 8 ans ; deux tiers des cas surviennent avant l’âge de 10 ans. 90 % des cas surviennent avant 16 ans, avec une légère prédominance masculine (ratio 5:3). Il n’y a pas de différence d’incidence selon la race. La tumeur est bien délimitée et siège le plus souvent dans la partie supérieure de l’orbite.
La répartition des sites de rhabdomyosarcome oculaire est : orbite 76 %, conjonctive 12 %, uvée 9 %, paupière 3 %. La tumeur peut naître dans l’orbite ou provenir de tissus environnants comme les sinus paranasaux et envahir l’orbite. L’orbite est le site primitif de 10 % de tous les rhabdomyosarcomes ; les autres sites incluent l’appareil urogénital (22 %), les membres (18 %), les régions paraméningées (16 %) et d’autres sites de la tête et du cou (10 %).
QÀ quel âge le rhabdomyosarcome survient-il le plus souvent ?
A
Les deux tiers des cas surviennent chez des enfants de moins de 10 ans, avec un âge moyen de 7 à 8 ans. 90 % des cas surviennent avant l’âge de 16 ans. L’apparition chez l’adulte est rare mais rapportée.
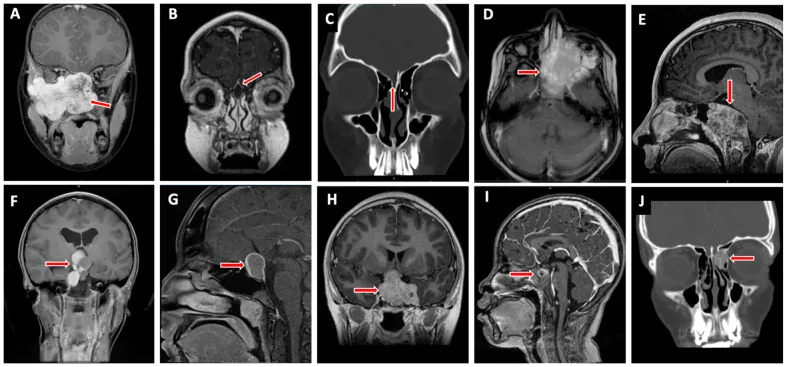
Valencia-Sanchez BA, et al. Special Considerations in Pediatric Endoscopic Skull Base Surgery. J Clin Med. 2024. Figure 2. PMCID: PMC11013018. License: CC BY.
La flèche rouge indique un rhabdomyosarcome (D), l’une des lésions typiques de la base du crâne chez l’enfant. Cela correspond à l’infiltration tumorale traitée dans la section « 2. Principaux symptômes et signes cliniques ».
Exophtalmie rapide : Une exophtalmie unilatérale progressant rapidement en quelques semaines est un symptôme typique.
Œdème palpébral : La paupière gonfle à mesure que la tumeur grossit. Même en cas d’œdème important, la rougeur est légère, les signes inflammatoires sont rares et la douleur n’est pas intense, ce qui est caractéristique.
Limitation des mouvements oculaires, ptosis, etc., présentant divers tableaux cliniques selon la localisation de la tumeur.
Survient fréquemment chez les enfants de 0 à 9 ans et se caractérise par une progression rapide.
Les lésions sont plus fréquentes dans les régions supérieure et supéro-interne, la masse déviant le globe oculaire vers le bas et l’extérieur. On observe une masse ronde à ovale, aux limites relativement nettes, en dehors du cône musculaire.
Environ 10 % des cas proviennent des sinus paranasaux et de la cavité nasale, envahissant secondairement l’orbite, et peuvent se manifester par une sinusite, une obstruction nasale ou une épistaxis. Les tumeurs postérieures peuvent s’accompagner de plis choroïdiens, de décollement de la rétine ou d’œdème papillaire.
Dans les cas primitifs de la conjonctive, la tumeur apparaît comme une masse granulomateuse rose en forme de grappe dans le cul-de-sac conjonctival. Dans les cas primitifs de l’uvée, elle se présente comme une masse irienne, pouvant s’accompagner d’ensemencement dans la chambre antérieure ou de glaucome secondaire.
Les métastases sont les plus fréquentes dans les poumons, suivies par la moelle osseuse, les os et les ganglions lymphatiques. L’orbite ne contient presque pas de vaisseaux lymphatiques, mais les tumeurs antérieures de la conjonctive et des paupières peuvent métastaser vers les ganglions lymphatiques régionaux.
QQuelles sont les maladies qui peuvent être confondues avec le rhabdomyosarcome ?
A
Les diagnostics différentiels cliniques incluent le neuroblastome, le chlorome, le lymphangiome, l’hémangiome infantile, la cellulite orbitaire et l’inflammation non spécifique. De plus, l’extension orbitaire d’une sinusite, la rupture d’un kyste dermoïde, le sarcome d’Ewing et l’infiltration orbitaire leucémique sont également inclus dans le diagnostic différentiel. La différenciation se fait par une progression rapide, l’imagerie et les résultats de biopsie.
Le rhabdomyosarcome ne provient pas des muscles extra-oculaires, mais de cellules mésenchymateuses indifférenciées pluripotentes dans les tissus mous orbitaires. La plupart des cas sont sporadiques et la cause exacte est inconnue.
Les facteurs de risque génétiques associés sont les suivants :
Une TDM orbitaire et crânienne en urgence est réalisée pour vérifier la présence d’une tumeur intra-orbitaire, son homogénéité, la destruction de la paroi orbitaire et l’extension intracrânienne ou sinusienne. Ensuite, une IRM est effectuée pour évaluer la nature interne de la tumeur et ses relations avec le globe oculaire et les muscles extra-oculaires.
Les résultats caractéristiques de chaque modalité sont les suivants :
Examen
Principaux résultats
TDM
Tumeur ronde à ovale, homogène, bien limitée. Rehaussement modéré à marqué.
IRM T1
Hypersignal par rapport à la graisse orbitaire, isosignal par rapport aux muscles extra-oculaires.
En IRM, la distinction avec un hémangiome capillaire peut être difficile, mais l’hémangiome capillaire est riche en vaisseaux (flux sanguin), ce qui permet de les différencier. Lorsque la composante stromale est importante, l’intensité du signal IRM diffère.
La recherche de métastases comprend une radiographie thoracique, une scintigraphie osseuse et une cytologie par ponction de moelle osseuse. La TEP/TDM, le scanner corps entier et la scintigraphie sont également utilisés pour le bilan d’extension.
Une biopsie est nécessaire pour le diagnostic définitif. La cytoponction à l’aiguille fine est insuffisante ; une biopsie-exérèse ou une biopsie incisionnelle est réalisée.
Biopsie-exérèse : choisie lorsque l’ablation chirurgicale est possible sans endommager les structures importantes.
Biopsie incisionnelle : choisie lorsque la tumeur est volumineuse et située dans l’orbite postérieure.
L’examen extemporané peut ne pas permettre de distinguer la tumeur d’autres tumeurs. Le principe est de réaliser une biopsie en urgence et de débuter précocement une chimiothérapie et une radiothérapie.
La mise en évidence de rhabdomyoblastes par microscopie optique, immunohistochimie et microscopie électronique est au cœur du diagnostic.
Les stries sont détectées par coloration HE ou trichrome de Masson. L’immunohistochimie est utilisée pour différencier les cellules indifférenciées : un profil desmin positif, HHF-35 (actine) positif, α-actine musculaire lisse négatif est utile au diagnostic. La microscopie électronique révèle des faisceaux d’actine-myosine et des protéines des bandes A, I et Z.
L’exérèse chirurgicale complète est impossible et ne doit pas être visée. Actuellement, le but principal de la chirurgie est d’obtenir un diagnostic tissulaire ; l’époque où l’exentération orbitaire était le premier choix est révolue. Après confirmation du diagnostic par biopsie-exérèse, un scanner thoraco-abdominal et une biopsie médullaire sont réalisés en pédiatrie pour rechercher des métastases, notamment pulmonaires. La chimiothérapie systémique est la base du traitement ; une trithérapie associant chirurgie, radiothérapie et chimiothérapie est le traitement standard.
La chimiothérapie utilise principalement le protocole VAC (vincristine + actinomycine D + cyclophosphamide). L’ifosfamide et l’étoposide ont également montré leur bénéfice.
La radiothérapie délivre environ 40 à 60 Gy. Depuis avril 2016, la protonthérapie est prise en charge par l’assurance maladie et s’ajoute aux options thérapeutiques standard. La protonthérapie présente l’avantage de réduire la dose aux tissus sains. La greffe de cellules souches hématopoïétiques est également pratiquée dans certains cas.
Le rhabdomyosarcome orbitaire est traité selon la classification de l’Intergroup Rhabdomyosarcoma Study (IRS). Dans une série de 30 cas de Shields et al., la répartition était : groupe I 7 %, groupe II 37 %, groupe III 53 %, groupe IV 3 %.
Groupe
Définition
Stratégie thérapeutique
I
Résection complète de la maladie locale
Chimiothérapie seule
II
Maladie résiduelle ou métastases ganglionnaires régionales
Chimiothérapie + radiothérapie
III
Résection incomplète ou résidu macroscopique
Chimiothérapie + radiothérapie
IV
Métastases à distance
Chimiothérapie + radiothérapie
Pour les groupes II à IV, la radiothérapie est administrée à une dose de 4000 à 5000 cGy (40 à 50 Gy) sur 4 à 5 semaines.
Pour les cas non résécables ou les récidives, une radiothérapie palliative ou une chimiothérapie peut être envisagée. Le traitement chirurgical peut inclure l’exérèse tumorale ou l’exentération orbitaire. Même en cas de résection complète, la chimiothérapie est obligatoire. L’orbite est un site de bon pronostic, avec une survie attendue de plus de 90 % si le traitement est approprié.
L’infiltration intracrânienne ou sinusienne, ou les métastases pulmonaires, systémiques ou ganglionnaires cervicales, sont associées à un mauvais pronostic vital. Un diagnostic précoce et un début précoce de la chimiothérapie améliorent la réponse et le pronostic vital.
Les complications ophtalmiques de la chimiothérapie comprennent la kérato-conjonctivite sèche et la blépharo-conjonctivite dues au cyclophosphamide, la conjonctivite et la vision trouble dues à l’ifosfamide, et l’occlusion de l’artère centrale de la rétine due à l’étoposide.
Les récidives locales sont évaluées par IRM régulière. Les métastases hématogènes sont fréquentes, nécessitant un bilan systémique régulier.
Les résultats visuels à long terme après traitement (Shields) sont : 20/20 à 20/40 dans 39 %, 20/50 à 20/100 dans 18 %, et 20/200 à absence de perception lumineuse dans 43 %.
QQuelles complications oculaires peuvent survenir après le traitement du rhabdomyosarcome ?
A
Les principales complications après radiothérapie (4000-5000 cGy) comprennent la rétinopathie radique (90 %), la cataracte radique (55 %), la sécheresse oculaire (36 %), l’hypoplasie orbitaire (24 %) et le ptosis (9 %). La chimiothérapie peut également provoquer une kérato-conjonctivite sèche, une conjonctivite ou une occlusion de l’artère centrale de la rétine selon les médicaments.
Selon la classification OMS, le rhabdomyosarcome est divisé en quatre sous-types : embryonnaire, alvéolaire, pléomorphe et à cellules fusiformes/sclérosant 2).
Type embryonnaire
Fréquence : représente 50 à 60 % de tous les rhabdomyosarcomes et 80 à 84 % des rhabdomyosarcomes orbitaires.
Histologie : faisceaux de cellules fusiformes (cellules en forme de fuseau) orientés dans diverses directions. Petites cellules rondes ou fusiformes avec peu de pléomorphisme nucléaire. Cytoplasme peu abondant avec des granules éosinophiles, et présence de rhabdomyoblastes striés. Plus fréquent dans la partie inférieure de l’orbite.
Pronostic : le taux de survie à 5 ans pour les cas orbitaires est de 95 %, ce qui est favorable. Âge moyen d’apparition : 7 à 8 ans.
Type alvéolaire
Fréquence : environ 20 % de tous les rhabdomyosarcomes. Environ 10 % des rhabdomyosarcomes orbitaires.
Histologie : prolifération tumorale en nids alvéolaires. Grandes cellules irrégulières avec un noyau volumineux et un cytoplasme abondant éosinophile, disposées autour de travées fibreuses. Tendance à des métastases étendues.
Pronostic : le taux de survie à 5 ans pour les cas orbitaires est de 74 %. C’est le type histologique le plus malin et de moins bon pronostic.
Type pléomorphe (différencié)
Fréquence : rare chez l’enfant. Survient principalement au niveau des membres (surtout la cuisse) chez l’adulte.
Histologie : cellules rondes à allongées, multinucléées avec un cytoplasme abondant, dans lesquelles on peut identifier des stries. Constitué de cellules tumorales très pléomorphes. Peu fréquent mais hautement différencié.
Pronostic : relativement favorable dans les cas orbitaires.
Le type botryoïde (botryoid) est une variante du type embryonnaire fréquente chez le nourrisson, caractérisée par des amas de cellules tumorales sous-épithéliaux donnant un aspect « en grappe de raisin ».
Sites de bon pronostic : orbite, appareil urogénital (sauf vessie et prostate), tête et cou (sauf sites paraméningés)
Sites de mauvais pronostic : membres, vessie, prostate, sites paraméningés
Histologiquement, les types alvéolaire et pléomorphe ont un pronostic plus défavorable que le type embryonnaire. Le taux de survie à 5 ans pour l’ensemble des rhabdomyosarcomes est de 66 %, mais le rhabdomyosarcome orbitaire est classé dans le groupe de bon pronostic en raison de sa localisation.
Le rhabdomyosarcome alvéolaire est caractérisé par des translocations chromosomiques récurrentes t(2;13)(q35;q14) et t(1;13)(p36;q14). Environ 80 % des rhabdomyosarcomes alvéolaires impliquent les translocations PAX3-FOXO1 ou PAX7-FOXO1. La fusion PAX3-FOXO1 est associée à une forte expression d’OLIG2 et à un mauvais pronostic2).
Le rhabdomyosarcome embryonnaire ne présente pas de réarrangements chromosomiques structurels récurrents, mais une perte allélique fréquente sur le chromosome 11 (en particulier la région 11p15.5) est observée.
QQuelles sont les différences de pronostic entre les types embryonnaire et alvéolaire ?
A
Dans le rhabdomyosarcome orbitaire, le taux de survie à 5 ans pour le type embryonnaire est de 95 %, tandis que pour le type alvéolaire, il est plus faible, à 74 %. Le type alvéolaire est associé à des translocations chromosomiques telles que PAX3-FOXO1 et est un type histologique sujet à des métastases étendues.
7. Recherches récentes et perspectives futures (rapports en phase de recherche)
Récemment, un nouveau sous-type appelé rhabdomyosarcome à cellules fusiformes épithélioïdes avec réarrangement TFCP2 a été identifié.
Li et al. (2023) ont rapporté que le rhabdomyosarcome à cellules fusiformes épithélioïdes survient fréquemment dans les os (tête et cou, bassin) et se caractérise par des fusions EWSR1-TFCP2 ou FUS-TFCP2, un sous-type au pronostic extrêmement défavorable3). La survie médiane rapportée n’est que de 17 mois.
Des recherches thérapeutiques ciblant la protéine de fusion PAX3-FOXO1, impliquée dans environ 80 % des rhabdomyosarcomes alvéolaires, sont en cours.
Un médicament contenant de petits ARN interférents (siARN) encapsulés dans des particules liposome-protamine a été rapporté pour réguler à la baisse efficacement l’expression de PAX3-FOXO1 dans des lignées cellulaires de rhabdomyosarcome alvéolaire in vitro et pour entraîner un retard et une suppression de la croissance des tumeurs de xénogreffe de rhabdomyosarcome alvéolaire1).
L’application d’inhibiteurs de points de contrôle immunitaires (tels que le nivolumab) aux sarcomes métastatiques est en cours d’essai1). Cependant, à l’heure actuelle, leur efficacité contre le rhabdomyosarcome n’est pas établie.
Traitement du rhabdomyosarcome primitif de l’adulte
Une revue de la littérature portant sur le rhabdomyosarcome alvéolaire primitif de la région pinéale chez l’adulte suggère une association entre l’intensité du traitement et la survie.
Dans une revue de 13 cas de rhabdomyosarcome alvéolaire primitif de la région pinéale chez l’adulte par Chang et al. (2025), la survie moyenne était d’environ 5 mois avec la chirurgie seule, d’environ 10,28 mois avec la chirurgie plus radiothérapie, et d’environ 11,33 mois avec la chirurgie plus radiothérapie plus chimiothérapie, montrant une tendance à une survie prolongée avec un traitement combiné2).
Yang N, Kong D, Wang X, Liu Y. Perianal rhabdomyosarcoma in an adult: A case report and review of the literature. Medicine. 2023;102(48):e36199.
Chang T, Ding C, Liu Y, Yang Y, Mao Q.. Primary pineal alveolar rhabdomyosarcoma in an adult patient: a case report and literature review. BMC Neurol. 2025;25(1):106. doi:10.1186/s12883-025-04113-8. PMID:40082819; PMCID:PMC11905731.
Li Y, Li D, Wang J, Tang J.. Epithelioid and spindle rhabdomyosarcoma with TFCP2 rearrangement in abdominal wall: a distinctive entity with poor prognosis. Diagn Pathol. 2023;18(1):41. doi:10.1186/s13000-023-01330-y. PMID:36998041; PMCID:PMC10061849.
Copiez le texte de l'article et collez-le dans l'assistant IA de votre choix.
Article copié dans le presse-papiers
Ouvrez un assistant IA ci-dessous et collez le texte copié dans la conversation.