Rhabdomyosarcoma is a malignant tumor derived from mesenchymal cells in the orbit, showing differentiation toward striated muscle. It arises from undifferentiated mesenchymal cells within the orbit and is not a malignant transformation of differentiated muscle tissue such as extraocular muscles. It is the most common malignant orbital tumor in children and should be the first consideration when a rapidly expanding orbital tumor is seen in a child.
In the United States, it accounts for about 5% of all childhood cancers, with approximately 250 new cases diagnosed annually. Of these, about 10% (about 35 cases per year) are primary orbital tumors. The annual incidence is estimated at 4 cases per million population overall and 4.5 cases per million in children 1).
The average age at onset is 7–8 years, with two-thirds occurring in children under 10 years. 90% occur under 16 years, with a slight male predominance (male-to-female ratio 5:3). There is no difference in incidence by race. The tumor is well-defined and often located in the upper orbit.
The distribution of orbital rhabdomyosarcoma by site is: orbit 76%, conjunctiva 12%, uvea 9%, and eyelid 3%. It may arise within the orbit or invade the orbit from surrounding tissues such as the paranasal sinuses. The orbit is the primary site in 10% of all rhabdomyosarcomas; other sites include the genitourinary tract (22%), extremities (18%), parameningeal region (16%), and other head and neck areas (10%).
QAt what age does rhabdomyosarcoma most commonly occur?
A
Two-thirds of cases occur in children under 10 years of age, with an average age of onset of 7–8 years. 90% occur in children under 16 years. Onset in adults is rare but has been reported.
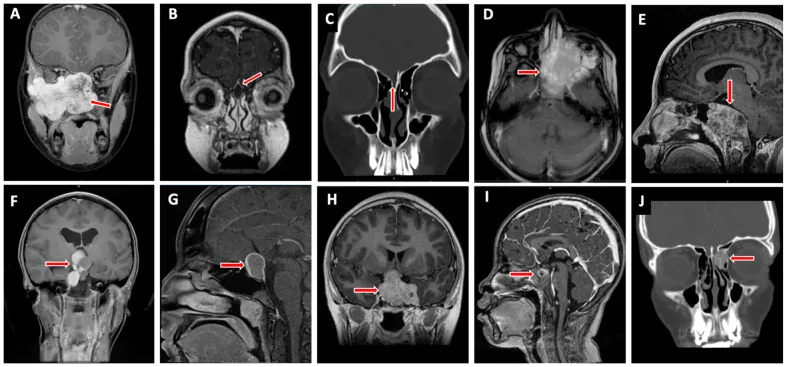
Valencia-Sanchez BA, et al. Special Considerations in Pediatric Endoscopic Skull Base Surgery. J Clin Med. 2024. Figure 2. PMCID: PMC11013018. License: CC BY.
The red arrow indicates rhabdomyosarcoma (D), one of the representative lesions at the skull base in children. This corresponds to the tumor infiltration discussed in the section “2. Main Symptoms and Clinical Findings”.
Rapid proptosis: Rapidly progressive unilateral proptosis over several weeks is a typical symptom.
Eyelid swelling: The eyelid swells as the tumor enlarges. Even with severe swelling, redness is mild, inflammatory findings are scarce, and pain is not severe, which is characteristic.
Various clinical presentations such as restricted eye movement and ptosis, depending on the tumor location.
It commonly occurs in children aged 0–9 years and is characterized by rapid progression.
Lesions are most commonly located superiorly or superomedially, displacing the eye downward and outward. A relatively well-circumscribed round to oval mass is found outside the muscle cone.
Approximately 10% of cases arise from the paranasal sinuses or nasal cavity and secondarily invade the orbit, which may present with sinusitis, nasal congestion, or epistaxis. Posterior tumors may be associated with choroidal folds, retinal detachment, or optic disc edema.
Conjunctival primary lesions appear as a pink, grape-like granulomatous mass in the conjunctival fornix. Uveal primary lesions present as iris masses and may be accompanied by anterior chamber seeding or secondary glaucoma.
Metastases most commonly occur in the lungs, followed by bone marrow, bone, and lymph nodes. Although the orbit has almost no lymphatic vessels, anterior tumors of the conjunctiva and eyelid can metastasize to regional lymph nodes.
QWhat diseases are easily mistaken for rhabdomyosarcoma?
A
Clinical differential diagnoses include neuroblastoma, chloroma, lymphangioma, infantile hemangioma, orbital cellulitis, and nonspecific inflammation. Additionally, orbital extension of sinusitis, dermoid cyst rupture, Ewing sarcoma, and orbital infiltration of leukemia are also included in the differential diagnosis. Differentiation is made by rapid progression, imaging findings, and biopsy results.
Rhabdomyosarcoma arises not from extraocular muscles but from undifferentiated mesenchymal cells with multilineage potential in the orbital soft tissues. Most cases are sporadic, and the exact cause is unknown.
Associated genetic risk factors are as follows:
Neurofibromatosis type 1: Known to be associated with an increased risk of developing rhabdomyosarcoma.
Li-Fraumeni syndrome: Involves abnormalities in the p53 tumor suppressor gene.
Beckwith-Wiedemann syndrome: A congenital disorder characterized by overgrowth, umbilical hernia, and hypoglycemia.
Hereditary retinoblastoma: May occur as a secondary cancer after radiation therapy.
Emergency head and orbital CT is performed to check for the presence of an intraorbital tumor, tumor homogeneity, destruction of the orbital bone wall, and extension into the intracranial space or paranasal sinuses. Next, MRI is performed to assess the internal characteristics of the tumor and its positional relationship with the eyeball and extraocular muscles.
Characteristic findings of each modality are as follows:
Examination
Main Findings
CT
Homogeneous, well-defined round to oval mass. Moderate to marked contrast enhancement.
MRI T1
Hypointense relative to orbital fat, isointense relative to extraocular muscles.
On MRI, differentiation from capillary hemangioma may be difficult, but capillary hemangioma can be distinguished by its rich vascularity (blood flow). When stromal components are abundant, MRI signal intensity differs.
For metastasis workup, chest X-ray, bone scintigraphy, and bone marrow aspiration cytology are performed. PET/CT, whole-body CT, and scintigraphy are also used for systemic evaluation.
Biopsy is necessary for definitive diagnosis. Fine-needle aspiration biopsy is insufficient; excisional or incisional biopsy should be performed.
Excisional biopsy: Selected when surgical removal is possible without damaging important structures.
Incisional biopsy: Selected when the tumor is large or located in the posterior orbit.
Intraoperative frozen section diagnosis may be difficult to distinguish from other tumors. The principle is to perform biopsy via emergency surgery and start chemotherapy/radiotherapy early.
Demonstration of rhabdomyoblasts by light microscopy, immunohistochemistry, and electron microscopy is central to diagnosis.
Striations are detected by HE staining and Masson trichrome staining. Immunohistochemical staining is used to differentiate undifferentiated cells, and a pattern of desmin-positive, HHF-35 (actin)-positive, and α-smooth muscle actin-negative is useful for diagnosis. Electron microscopy reveals actin-myosin bundles and A, I, and Z band proteins.
Clinical differential diagnoses: neuroblastoma, chloroma, lymphangioma, infantile hemangioma, orbital cellulitis, nonspecific inflammation. Additionally, orbital extension of sinusitis, orbital cellulitis, dermoid cyst rupture, intratumoral hemorrhage of hemangioma, hemorrhage of lymphangioma, Ewing sarcoma, orbital infiltration of leukemia (chloroma), and orbital hemorrhage are also considered in the differential.
Complete surgical resection is not possible and should not be attempted. Currently, the main purpose of surgery is to obtain a tissue diagnosis, and the era of orbital exenteration as the first choice has ended. After confirming the pathological diagnosis by excisional biopsy, chest and abdominal CT and bone marrow biopsy are performed in the pediatrics department to check for systemic metastases such as to the lungs. Systemic chemotherapy is the mainstay of treatment, and a combination of surgery, radiotherapy, and chemotherapy is the standard treatment.
For chemotherapy, VAC therapy (vincristine + actinomycin D + cyclophosphamide) is mainly used. The benefits of ifosfamide and etoposide have also been reported.
Radiotherapy is administered at approximately 40–60 Gy. Since April 2016, proton beam therapy has been covered by insurance and has become a standard treatment option. Proton beam therapy has the advantage of reducing the dose to normal tissues. Hematopoietic stem cell transplantation is also performed in some cases.
The treatment strategy for orbital rhabdomyosarcoma is determined based on the Intergroup Rhabdomyosarcoma Study (IRS) staging system. In a series of 30 cases by Shields et al., the distribution was Group I 7%, Group II 37%, Group III 53%, and Group IV 3%.
Group
Definition
Treatment Strategy
I
Complete resection of localized disease
Chemotherapy alone
II
Residual disease or regional lymph node metastasis
Chemotherapy + radiation therapy
III
Incomplete resection or gross residual disease
Chemotherapy + radiation therapy
IV
Distant metastasis
Chemotherapy + radiation therapy
For groups II to IV, radiation therapy is administered at 4000–5000 cGy (40–50 Gy) over 4–5 weeks.
For cases that cannot be completely resected or for recurrence, palliative treatment such as radiation therapy or chemotherapy may be considered. Surgical treatments include tumor resection and orbital exenteration. Even if complete resection is achieved surgically, chemotherapy is mandatory. The orbit is a favorable prognostic site, and with appropriate treatment, a survival rate of over 90% can be expected.
If there is invasion into the intracranial cavity or paranasal sinuses, or metastasis to the lungs, other parts of the body, or cervical lymph nodes, the prognosis is poor. Early detection and early initiation of chemotherapy lead to a good response and improved prognosis.
Ophthalmic complications of chemotherapy include dry keratoconjunctivitis and blepharoconjunctivitis due to cyclophosphamide, conjunctivitis and blurred vision due to ifosfamide, and central retinal artery occlusion due to etoposide.
Local recurrence is evaluated by regular MRI examinations. Hematogenous metastasis is common, so regular systemic examinations are performed.
Long-term visual outcomes after treatment (Shields) are: 20/20 to 20/40 in 39%, 20/50 to 20/100 in 18%, and 20/200 to no light perception in 43%.
QWhat eye complications can occur after treatment for rhabdomyosarcoma?
Rhabdomyosarcoma is classified by the WHO into four subtypes: embryonal, alveolar, pleomorphic, and spindle cell/sclerosing 2).
Embryonal type
Frequency: Accounts for 50–60% of all rhabdomyosarcomas and 80–84% of orbital rhabdomyosarcomas.
Histology: Bundles of spindle-shaped cells (villous cells) running in various directions. Cells are small round or spindle-shaped with minimal nuclear pleomorphism. The cytoplasm is scant with eosinophilic granules, and rhabdomyoblasts with cross-striations are present. More common in the lower orbit.
Prognosis: The 5-year survival rate for orbital cases is 95%, which is favorable. Mean age at onset is 7–8 years.
Alveolar Type
Frequency: Accounts for about 20% of all rhabdomyosarcomas and about 10% of orbital rhabdomyosarcomas.
Histology: The tumor grows in an alveolar pattern. Large, irregular cells with large nuclei and abundant eosinophilic cytoplasm are arranged around fibrous septa. Prone to widespread metastasis.
Prognosis: The 5-year survival rate for orbital cases is 74%. This is the most malignant and prognostically unfavorable histological type.
Pleomorphic Type (Differentiated Type)
Frequency: Rare in children. Mainly occurs in the extremities (especially the thigh) of adults.
Histology: Round to elongated cells with multinucleation and abundant cytoplasm, with identifiable cross-striations. Composed of highly pleomorphic tumor cells. Low frequency but high differentiation.
Prognosis: Relatively favorable in orbital cases.
The botryoid type is a variant of embryonal type common in infants, characterized by subepithelial tumor cell aggregates giving a “grape-like” appearance.
Histologically, the alveolar and pleomorphic types have a poorer prognosis than the embryonal type. The overall 5-year survival rate for rhabdomyosarcoma is 66%, but orbital rhabdomyosarcoma is classified as a favorable prognosis group based on its site of origin.
Alveolar rhabdomyosarcoma is characterized by recurrent chromosomal translocations t(2;13)(q35;q14) and t(1;13)(p36;q14). Approximately 80% of alveolar rhabdomyosarcomas involve PAX3-FOXO1 or PAX7-FOXO1 translocations. PAX3-FOXO1 fusion is associated with high OLIG2 expression and poor prognosis2).
Embryonal rhabdomyosarcoma does not have recurrent structural chromosomal rearrangements, but allelic loss on chromosome 11 (especially the 11p15.5 region) is frequently observed.
QWhat is the difference in prognosis between embryonal and alveolar types?
A
The 5-year survival rate for embryonal type in orbital rhabdomyosarcoma is 95%, whereas for alveolar type it is lower at 74%. The alveolar type is associated with chromosomal translocations such as PAX3-FOXO1 and is a histological type prone to widespread metastasis.
7. Latest Research and Future Perspectives (Investigational Reports)
In recent years, a new subtype called epithelioid spindle cell rhabdomyosarcoma with TFCP2 rearrangement has been identified.
Li et al. (2023) reported that epithelioid spindle cell rhabdomyosarcoma preferentially occurs in bones (head and neck, pelvis) and is characterized by EWSR1-TFCP2 or FUS-TFCP2 fusions, representing an extremely poor prognosis subtype3). The median survival of reported cases is only 17 months.
Therapeutic research targeting the PAX3-FOXO1 fusion protein, which is involved in approximately 80% of alveolar rhabdomyosarcomas, is ongoing.
A formulation encapsulating small interfering RNA (siRNA) in liposome-protamine particles has been reported to efficiently downregulate PAX3-FOXO1 expression in alveolar rhabdomyosarcoma cell lines in vitro and to delay or suppress the growth of alveolar rhabdomyosarcoma xenograft tumors1).
Immune checkpoint inhibitors (such as nivolumab) are being investigated for use in metastatic sarcoma1). However, their efficacy in rhabdomyosarcoma has not been established at this time.
A literature review of adult primary alveolar rhabdomyosarcoma of the pineal region suggests an association between treatment intensity and survival duration.
In a review of 13 cases of adult primary alveolar rhabdomyosarcoma of the pineal region by Chang et al. (2025), the mean survival was approximately 5 months with surgery alone, about 10.28 months with surgery plus radiotherapy, and about 11.33 months with surgery plus radiotherapy plus chemotherapy, indicating a trend toward prolonged survival with multimodal therapy2).
Yang N, Kong D, Wang X, Liu Y. Perianal rhabdomyosarcoma in an adult: A case report and review of the literature. Medicine. 2023;102(48):e36199.
Chang T, Ding C, Liu Y, Yang Y, Mao Q.. Primary pineal alveolar rhabdomyosarcoma in an adult patient: a case report and literature review. BMC Neurol. 2025;25(1):106. doi:10.1186/s12883-025-04113-8. PMID:40082819; PMCID:PMC11905731.
Li Y, Li D, Wang J, Tang J.. Epithelioid and spindle rhabdomyosarcoma with TFCP2 rearrangement in abdominal wall: a distinctive entity with poor prognosis. Diagn Pathol. 2023;18(1):41. doi:10.1186/s13000-023-01330-y. PMID:36998041; PMCID:PMC10061849.
Copy the article text and paste it into your preferred AI assistant.
Article copied to clipboard
Open an AI assistant below and paste the copied text into the chat box.