Rabdomyosarkom, orbital mezenkimal hücrelerden kaynaklanan ve çizgili kasa farklılaşma gösteren malign bir tümördür. Orbita içindeki farklılaşmamış mezenkimal hücrelerden gelişir; ekstraoküler kaslar gibi farklılaşmış kas dokusunun malignleşmesi sonucu oluşmaz. Çocuklarda en sık görülen orbital malign tümördür ve çocukta hızla büyüyen orbital tümör görüldüğünde akla gelmesi gereken ilk hastalıktır.
ABD’de tüm çocukluk çağı kanserlerinin yaklaşık %5’ini oluşturur ve yılda yaklaşık 250 yeni vaka tanı alır. Bunların yaklaşık %10’u (yılda yaklaşık 35 vaka) orbital kaynaklıdır. Yıllık insidans tüm popülasyonda 4/1.000.000, çocuklarda ise 4,5/1.000.000 olarak tahmin edilmektedir1).
Ortalama görülme yaşı 7-8 olup, üçte ikisi 10 yaş altı çocuklarda görülür. %90’ı 16 yaş altında ortaya çıkar ve erkeklerde biraz daha sıktır (kız/erkek oranı 5:3). Irklar arasında insidans farkı yoktur. Tümör genellikle sınırları belirgindir ve orbitanın üst kısmında daha sıktır.
Oküler rabdomyosarkomun yerleşim yeri dağılımı: orbita %76, konjonktiva %12, uvea %9, göz kapağı %3’tür. Orbita içinde oluşabileceği gibi, paranazal sinüsler gibi çevre dokularda oluşan tümörün orbitaya invaze olmasıyla da görülebilir. Orbita, tüm rabdomyosarkomların %10’unun primer bölgesidir; diğer yerleşim yerleri arasında genitoüriner sistem (%22), ekstremiteler (%18), parameningeal bölge (%16) ve diğer baş-boyun bölgeleri (%10) bulunur.
QRabdomyosarkom en sık hangi yaşta görülür?
A
Üçte ikisi 10 yaş altı çocuklarda görülür ve ortalama başlangıç yaşı 7-8’dir. %90’ı 16 yaş altında ortaya çıkar. Erişkinlerde görülmesi nadirdir ancak bildirilmiştir.
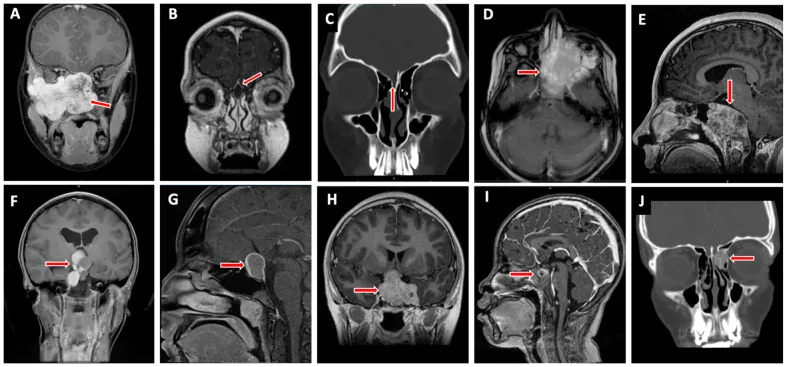
Valencia-Sanchez BA, et al. Special Considerations in Pediatric Endoscopic Skull Base Surgery. J Clin Med. 2024. Figure 2. PMCID: PMC11013018. License: CC BY.
Kırmızı ok, çocuklarda kafa tabanının yaygın lezyonlarından biri olan rabdomyosarkomu (D) göstermektedir. Bu görüntü, metnin «2. Ana belirtiler ve klinik bulgular» bölümünde ele alınan tümör invazyonuna karşılık gelmektedir.
Hızlı göz protrüzyonu: Birkaç hafta içinde hızla ilerleyen tek taraflı göz protrüzyonu tipik bir belirtidir.
Göz kapağı şişliği: Tümör büyüdükçe göz kapağı şişer. Şiddetli şişlik olsa bile kızarıklık hafiftir, inflamasyon bulguları azdır ve ağrı belirgin değildir; bu özellikler karakteristiktir.
Göz hareketlerinde kısıtlılık, pitozis gibi tümörün yerine bağlı çeşitli klinik tablolar görülür.
0-9 yaş arası çocuklarda sık görülür ve hızlı ilerleme karakteristiktir.
Shields ve arkadaşlarının raporuna göre bulguların sıklığı aşağıdaki gibidir:
Bulgular
Sıklık
Göz protrüzyonu
%80-100
Göz kayması
%80
Konjonktiva ve göz kapağı şişliği
%60
Göz kapağı düşüklüğü
%30-50
Ele gelen kitle
%25
Ağrı
%10
Lezyon çoğunlukla üst ve üst-iç kısımda yer alır ve kitle gözü aşağı ve dışa doğru kaydırır. Kas konisi dışında, sınırları nispeten belirgin, yuvarlak ila oval bir kitle görülür.
Yaklaşık %10 olguda paranazal sinüsler ve burun boşluğundan kaynaklanarak ikincil olarak yörüngeye invazyon olur ve sinüzit, burun tıkanıklığı ve burun kanaması görülebilir. Arka yerleşimli tümörlerde koroid kıvrımları, retina dekolmanı ve optik disk ödemi eşlik edebilir.
Konjonktiva kaynaklı olgularda, konjonktival fornikste granüler, pembe, üzüm salkımı benzeri bir kitle olarak ortaya çıkar. Uvea kaynaklı olgularda ise iris kitlesi olarak görülür ve ön kamara yayılımı ile sekonder glokom eşlik edebilir.
Metastazlar en sık akciğere, ardından sırasıyla kemik iliği, kemik ve lenf düğümlerine olur. Yörüngede lenf damarları neredeyse yoktur, ancak konjonktiva ve göz kapağının ön yerleşimli tümörleri bölgesel lenf düğümlerine metastaz yapabilir.
QRabdomyosarkom ile karıştırılabilecek hastalıklar nelerdir?
A
Klinik ayırıcı tanılar arasında nöroblastom, kloroma, lenfanjiyom, infantil hemanjiyom, orbital selülit ve nonspesifik inflamasyon yer alır. Ayrıca sinüzitin orbitaya yayılımı, dermoid kist rüptürü, Ewing sarkomu ve löseminin orbitaya infiltrasyonu da ayırıcı tanıya dahildir. Hızlı ilerleme, görüntüleme bulguları ve biyopsi ile ayırt edilir.
Rabdomiyosarkom, ekstraoküler kaslardan değil, orbital yumuşak dokudaki multipotent farklılaşmamış mezenkimal hücrelerden kaynaklanır. Çoğu sporadiktir ve net bir nedeni yoktur.
Acil olarak baş ve orbital BT çekilir; orbitada tümör olup olmadığı, tümör içinin homojen olup olmadığı, orbital kemik duvarında yıkım olup olmadığı, intrakraniyal ve sinüslere yayılım kontrol edilir. Ardından MRG yapılarak tümör iç özellikleri, göz küresi ve ekstraoküler kaslarla ilişkisi değerlendirilir.
Her modalitenin karakteristik bulguları aşağıdaki gibidir:
Test
Ana Bulgular
BT
Homojen, sınırları belirgin, yuvarlak-oval kitle. Orta-yüksek kontrast tutulumu
MRG T1
Orbita yağına göre düşük sinyal, ekstraoküler kaslara göre eşit sinyal
MRG T2
Orbita yağı ve ekstraoküler kaslara göre yüksek sinyal
MRG’de bazen kapiller hemanjiyomdan ayırt etmek zor olabilir, ancak kapiller hemanjiyom bol damar (kan akımı) içerdiğinden ayrım mümkündür. Stromal bileşenler fazlaysa MRG sinyal yoğunluğu farklı olur.
Metastaz taraması için akciğer grafisi, kemik sintigrafisi ve kemik iliği aspirasyon sitolojisi yapılır. Sistemik taramada PET/BT, tüm vücut BT ve sintigrafi de kullanılır.
Kesin tanı için biyopsi gereklidir. İnce iğne aspirasyon biyopsisi yetersizdir; eksizyonel veya insizyonel biyopsi yapılır.
Eksizyonel biyopsi: Önemli yapılara zarar vermeden cerrahi olarak çıkarılması mümkün olduğunda tercih edilir.
İnsizyonel biyopsi: Tümör büyük ve arka orbitada yerleşmişse tercih edilir.
İntraoperatif frozen kesit incelemesinde diğer tümörlerden ayırt etmek bazen zordur. Acil biyopsi yapılıp erken kemoterapi ve radyoterapi başlanması prensiptir.
Tanı, ışık mikroskopisi, immünohistokimya ve elektron mikroskopisi ile rabdomiyoblastların gösterilmesine dayanır.
HE ve Masson trikrom boyaması ile çizgilenme tespit edilir. Farklılaşmamış hücrelerin ayırıcı tanısı için immünohistokimyasal boyama yapılır ve desmin pozitif, HHF-35 (aktin) pozitif, α-düz kas aktin negatif paterni tanıda faydalıdır. Elektron mikroskopisinde aktin-miyozin demetleri, A, I ve Z bant proteinleri görülür.
Klinik ayırıcı tanılar: nöroblastom, kloroma, lenfanjiyom, infantil hemanjiyom, orbital selülit, nonspesifik inflamasyon. Ayrıca sinüzitin orbitaya yayılımı, orbital selülit, dermoid kist rüptürü, hemanjiyomda intratümöral kanama, lenfanjiyomda kanama, Ewing sarkomu, löseminin orbitaya infiltrasyonu (kloroma) ve orbital kanama da ayırıcı tanıda yer alır.
Tümörün cerrahi olarak tamamen çıkarılması mümkün değildir ve hedeflenmemelidir. Günümüzde cerrahinin ana amacı doku tanısı sağlamaktır ve orbital eksenterasyonun birinci seçenek olduğu dönem sona ermiştir. Eksizyonel biyopsi ile patolojik tanı konulduktan sonra, pediatri bölümünde torakoabdominal BT ve kemik iliği biyopsisi gibi tetkiklerle akciğer gibi uzak metastazlar araştırılır. Sistemik kemoterapi tedavinin temelidir ve cerrahi, radyoterapi ve kemoterapinin kombinasyonu standart tedavidir.
Kemoterapide başlıca VAC rejimi (vinkristin + aktinomisin D + siklofosfamid) kullanılır. Ayrıca ifosfamid ve etoposidin faydası da bildirilmiştir.
Radyoterapi yaklaşık 40-60 Gy dozunda uygulanır. Nisan 2016’dan itibaren proton tedavisi sigorta kapsamına alınmıştır ve standart tedavi seçeneklerine eklenmiştir. Proton tedavisi, normal dokuya verilen dozu azaltma avantajına sahiptir. Bazı vakalarda hematopoietik kök hücre nakli de yapılmaktadır.
Orbital rabdomiyosarkom, Intergroup Rhabdomyosarcoma Study (IRS) evrelemesine göre tedavi edilir. Shields ve arkadaşlarının 30 vakalık serisinde evre dağılımı: Grup I %7, Grup II %37, Grup III %53, Grup IV %3 idi.
Grup
Tanım
Tedavi Yaklaşımı
I
Lokal hastalığın tam rezeksiyonu
Sadece kemoterapi
II
Rezidüel hastalık veya bölgesel lenf nodu metastazı
Kemoterapi + Radyoterapi
III
Tam olmayan rezeksiyon veya makroskopik rezidü
Kemoterapi + Radyoterapi
IV
Uzak metastaz
Kemoterapi + Radyoterapi
Grup II ila IV için radyoterapi, 4-5 hafta boyunca 4000-5000 cGy (40-50 Gy) olarak uygulanır.
Tam rezeksiyon yapılamayan veya nüks eden olgularda palyatif tedavi olarak radyoterapi veya kemoterapi düşünülür. Cerrahi tedavi olarak tümör çıkarılması ve orbita ekzenterasyonu da yapılabilir. Cerrahi ile tam rezeksiyon yapılsa bile kemoterapi zorunludur. Orbita, prognozu iyi olan bir bölgedir ve uygun tedavi ile %90’ın üzerinde sağkalım beklenir.
Tümör kafa içine veya sinüslere invaze olmuşsa veya akciğer gibi vücudun diğer bölgelerine veya servikal lenf nodlarına metastaz yapmışsa hayati prognoz kötüdür. Erken tanı ve erken kemoterapi başlanması, kemoterapiye yanıtı ve hayati prognozu iyileştirir.
Kemoterapinin oküler komplikasyonları olarak, siklofosfamid kaynaklı kuru keratokonjonktivit ve blefarokonjonktivit, ifosfamid kaynaklı konjonktivit ve bulanık görme, etoposid kaynaklı retinal santral arter tıkanıklığı bildirilmiştir.
Lokal nüks, düzenli MRG incelemeleri ile değerlendirilir. Hematojen metastaz sık görülür ve düzenli sistemik muayeneler yapılır.
Tedavi sonrası uzun dönem görme sonuçları (Shields): 20/20-20/40 %39, 20/50-20/100 %18, 20/200-ışık hissi yok %43.
QRabdomyosarkom tedavisinden sonra hangi göz komplikasyonları ortaya çıkabilir?
A
Radyoterapi (4000-5000 cGy) sonrası başlıca komplikasyonlar olarak radyasyon retinopatisi (%90), radyasyon kataraktı (%55), kuru göz (%36), orbital hipoplazi (%24) ve pitozis (%9) bildirilmiştir. Kemoterapi de ilaçlara bağlı olarak kuru keratokonjonktivit, konjonktivit ve retinal santral arter tıkanıklığına neden olabilir.
Rabdomyosarkom, WHO sınıflamasına göre embriyonel, alveolar, pleomorfik ve iğsi hücreli/sklerozan olmak üzere dört alt tipe ayrılır2).
Embriyonel tip
Sıklık: Tüm rabdomyosarkomların %50-60’ını, orbital rabdomyosarkomların %80-84’ünü oluşturur.
Histoloji: Farklı yönlerde uzanan iğsi hücre (villöz hücre) demetleri. Küçük yuvarlak veya iğsi, nükleer pleomorfizm az. Sitoplazma az, eozinofilik granüller içerir ve çizgili rabdomiyoblastlar görülür. Orbita alt kısmında daha sık.
Prognoz: Orbital vakalarda 5 yıllık sağkalım %95 ile iyidir. Ortalama başlangıç yaşı 7-8.
Alveoler tip
Sıklık: Tüm rabdomyosarkomların yaklaşık %20’si. Orbital rabdomyosarkomların yaklaşık %10’u.
Histoloji: Tümör alveoler şekilde büyür. Büyük çekirdekli ve bol eozinofilik sitoplazmalı düzensiz büyük hücreler trabeküller etrafında dizilir. Yaygın metastaz yapma eğilimindedir.
Prognoz: Orbital vakalarda 5 yıllık sağkalım %74. En agresif ve en kötü prognozlu histolojik tiptir.
Pleomorfik (diferansiye) tip
Sıklık: Çocuklarda nadir. Esas olarak erişkinlerin ekstremitelerinde (özellikle uyluk) görülür.
Histoloji: Bol sitoplazmalı, çok çekirdekli yuvarlak ila uzun hücreler, çizgilenme ayırt edilebilir. Yüksek pleomorfizm gösteren tümör hücrelerinden oluşur. Sıklığı düşük ancak diferansiyasyon derecesi yüksektir.
Prognoz: Orbital vakalarda nispeten iyidir.
Botrioid tip, bebeklerde daha sık görülen embriyonel bir varyanttır ve subepitelyal tümör hücre agregatları ile karakterize olup ‘üzüm salkımı’ benzeri bir görünüm oluşturur.
İyi prognozlu bölgeler: Orbita, mesane ve prostat dışı ürogenital sistem, parameningeal olmayan baş-boyun
Kötü prognozlu bölgeler: Ekstremiteler, mesane, prostat, parameningeal
Histolojik olarak, alveolar ve pleomorfik tiplerin prognozu fetal tipe göre daha kötüdür. Tüm rabdomyosarkom için 5 yıllık sağkalım oranı %66 iken, orbital rabdomyosarkom oluşum yeri nedeniyle iyi prognozlu grupta sınıflandırılır.
Alveolar rabdomyosarkom, tekrarlayan kromozomal translokasyonlar t(2;13)(q35;q14) ve t(1;13)(p36;q14) ile karakterizedir. Alveolar rabdomyosarkomların yaklaşık %80’inde PAX3-FOXO1 veya PAX7-FOXO1 translokasyonu bulunur. PAX3-FOXO1 füzyonu, yüksek OLIG2 ekspresyonu ve kötü prognoz ile ilişkilidir2).
Fetal rabdomyosarkomda tekrarlayan yapısal kromozomal yeniden düzenlenmeler yoktur, ancak kromozom 11’de (özellikle 11p15.5 bölgesi) sık allel kaybı görülür.
QFetal ve alveolar tipler arasında prognoz açısından ne fark vardır?
A
Orbital rabdomyosarkomda fetal tip için 5 yıllık sağkalım oranı %95 iken, alveolar tip için %74 ile daha düşüktür. Alveolar tip, PAX3-FOXO1 gibi kromozomal translokasyonlarla birlikte görülür ve yaygın metastaza yatkın bir histolojik tiptir.
7. Güncel Araştırmalar ve Gelecek Perspektifleri (Araştırma Aşamasındaki Raporlar)
Son yıllarda, TFCP2 yeniden düzenlenmesi ile birlikte epiteloid iğsi hücreli rabdomyosarkom adı verilen yeni bir alt tip tanımlanmıştır.
Li ve ark. (2023), epiteloid iğsi hücreli rabdomyosarkomun kemiklerde (baş-boyun, pelvis) sık görüldüğünü ve EWSR1-TFCP2 veya FUS-TFCP2 füzyonu ile karakterize, son derece kötü prognozlu bir alt tip olduğunu bildirmiştir3). Bildirilen vakaların medyan sağkalımı sadece 17 aydır.
Alveolar rabdomyosarkomların yaklaşık %80’inde rol oynayan PAX3-FOXO1 füzyon proteinini hedef alan tedavi araştırmaları devam etmektedir.
Küçük enterferans yapan RNA (siRNA) içeren lipozom-protamin partikül formülasyonunun, in vitro olarak alveolar rabdomyosarkom hücre hatlarında PAX3-FOXO1 ekspresyonunu etkili bir şekilde aşağı regüle ettiği ve alveolar rabdomyosarkom ksenograft tümörlerinde büyüme gecikmesi ve baskılanması sağladığı bildirilmiştir1).
Metastatik sarkomda immün kontrol noktası inhibitörlerinin (nivolumab gibi) kullanımı araştırılmaktadır1). Ancak şu anda rabdomyosarkomda etkinliği kanıtlanmamıştır.
Erişkinlerde pineal bölge primer alveolar rabdomyosarkomu üzerine yapılan bir literatür taramasında, tedavi yoğunluğu ile sağkalım süresi arasında ilişki olduğu öne sürülmüştür.
Chang ve ark. (2025) tarafından erişkinlerde pineal bölge primer alveolar rabdomyosarkomlu 13 hastanın incelendiği bir derlemede, sadece cerrahi ile ortalama sağkalım yaklaşık 5 ay iken, cerrahi + radyoterapi ile yaklaşık 10.28 ay, cerrahi + radyoterapi + kemoterapi ile yaklaşık 11.33 ay olarak bildirilmiş ve çoklu tedavi kombinasyonu ile sağkalımın uzadığı gözlenmiştir2).
Yang N, Kong D, Wang X, Liu Y. Perianal rhabdomyosarcoma in an adult: A case report and review of the literature. Medicine. 2023;102(48):e36199.
Chang T, Ding C, Liu Y, Yang Y, Mao Q.. Primary pineal alveolar rhabdomyosarcoma in an adult patient: a case report and literature review. BMC Neurol. 2025;25(1):106. doi:10.1186/s12883-025-04113-8. PMID:40082819; PMCID:PMC11905731.
Li Y, Li D, Wang J, Tang J.. Epithelioid and spindle rhabdomyosarcoma with TFCP2 rearrangement in abdominal wall: a distinctive entity with poor prognosis. Diagn Pathol. 2023;18(1):41. doi:10.1186/s13000-023-01330-y. PMID:36998041; PMCID:PMC10061849.
Makale metnini kopyalayıp tercih ettiğiniz yapay zeka asistanına yapıştırabilirsiniz.
Makale panoya kopyalandı
Aşağıdaki yapay zeka asistanlarından birini açın ve kopyalanan metni sohbet kutusuna yapıştırın.